
Virome Profiling, New Virus Identification and the Prevalence and Distribution of Viruses Infecting Chieh-Qua (Benincasa hispida Cogn. var. chieh-qua How) in China
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
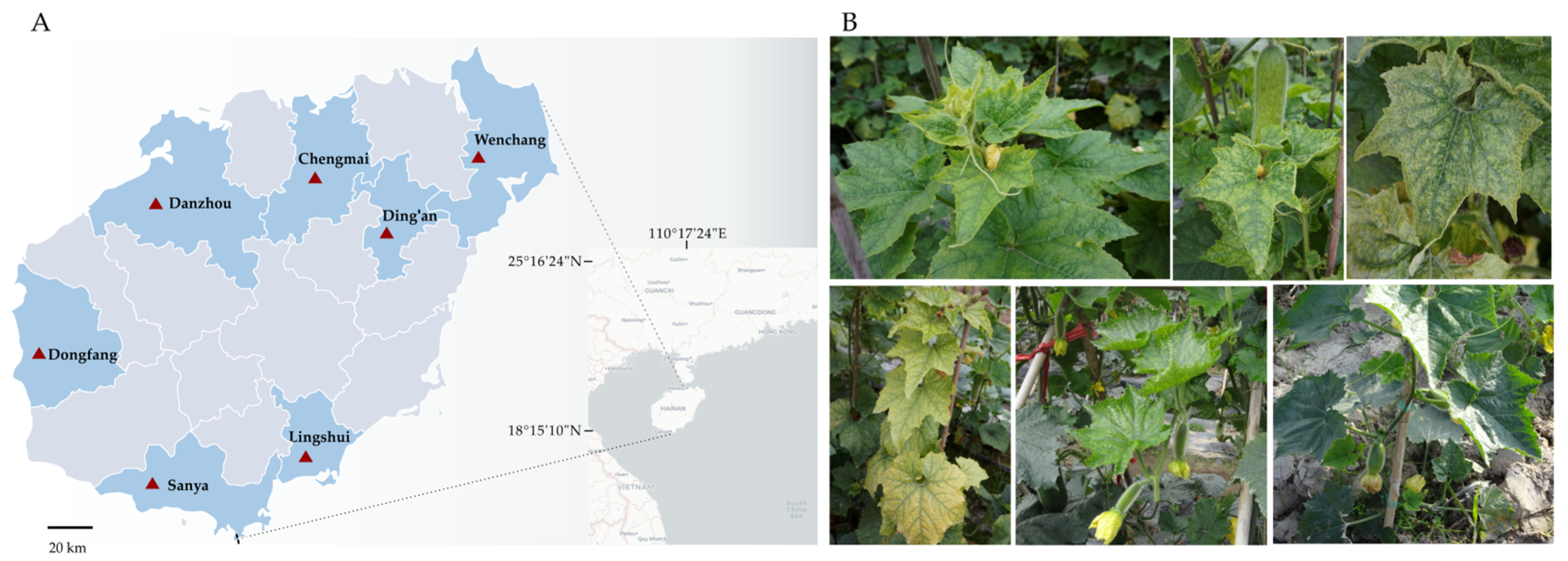
2.1. Plant Materials
2.2. Nucleic Acid Preparation and HTS
2.3. Data Processing and Virus Annotation
2.4. RT-PCR Protocol
2.5. Recovery of the Complete Genome and Characterization of the Newly Identified Viruses
2.6. Recombination Analysis
3. Results
3.1. HTS Output, Discovery of Viruses and HTS Validation
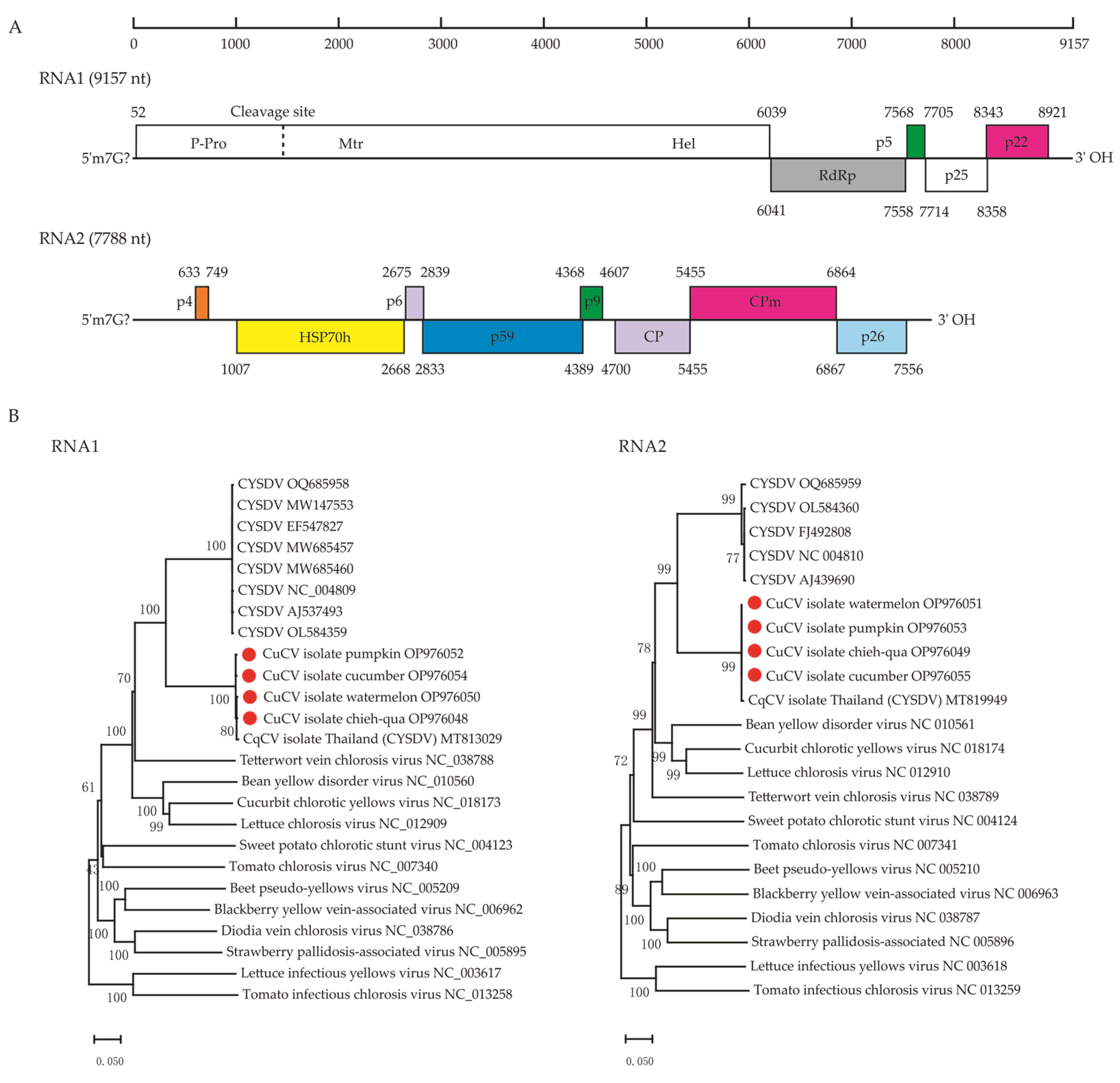
3.2. Recovery of the Complete Genomes and Characterization of a Potential New Crinivirus in the Family Closteroviridae
3.3. The Genetic Variability and Recombination of CuCV Isolates from Cucurbit Vegetables
3.4. A New Alphaendornavirus in Family Endornaviridae
3.5. The Prevalence of Viruses Infecting Chieh-Qua
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mao, Y.Z.; Jiang, B.; Peng, Q.W.; Liu, W.R.; Lin, Y.; Xie, D.S.; He, X.M.; Li, S.S. Cloning and characterization of WRKY gene homologs in Chieh-qua (Benincasa hispida Cogn. var. Chieh-qua How) and their expression in response to fusaric acid treatment. 3 Biotech 2017, 7, 86–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Department of Agriculture and Rural Affairs of Guangdong Province. Available online: http://dara.gd.gov.cn/nyyw/content/post_3327295.html (accessed on 15 April 2023).
- He, X.M.; Xie, D.S.; Chen, Q.H.; Peng, Q.W. Chieh-qua biotechnology, progress and prospects. Asian Australas. J. Plant Sci. Biotechnol. 2007, 1, 19–22. [Google Scholar]
- Zhou, S.J.; Chen, X.J.; Zhu, Y.Q.; Chen, L.P.; Zhang, P. Research progress and suggestion on germplasms of ash gourd and chieh-qua. J. Plant Genet. Resour. 2014, 15, 211–214. [Google Scholar]
- He, X.M.; Peng, Q.W.; Wang, M.; Yan, J.Q. Research Progresses in genetic breeding of chieh-qua in China. Guangdong Agri. Sci. 2021, 48, 1–11. [Google Scholar]
- Lu, C.G.; Li, H.F.; Fan, Z.F. Identification and cloning of coat protein gene of zucchini yellow mosaic virus infecting hairy squash. In Proceedings of the Annual Meeting of Chinese Society for Plant Pathology 2004, Ningbo, China, 1–4 September 2004; pp. 224–226. [Google Scholar]
- Zhou, C.J.; Liang, Z.R.; Zhang, J.B.; Huang, B.; He, G.Z.; Zhong, C.N.; Ouyang, T.X. First report of zucchini tigre mosaic virus infecting four cucurbit crops in China. Plant Dis. 2023, 107, 1247. [Google Scholar] [CrossRef]
- Zhao, Q.; Li, H.P.; Xie, D.S.; Luo, S.B.; Peng, Q.W. Development of a multiplex RT-PCR protocol for the detection of three viruses on Benincasa hispida. var. chieh-qua. Acta Hortic. Sin. 2011, 38, 2215–2222. [Google Scholar]
- Liu, Y.; Li, F.; Li, Y.Y.; Zhang, S.B.; Gao, X.W.; Xie, Y.; Yan, F.; Zhang, A.S.; Dai, L.Y.; Cheng, Z.B.; et al. Identification, distribution and occurrence of viruses in the main vegetables of China. Sci. Agri. Sin. 2019, 52, 239–261. [Google Scholar]
- Xiao, L.; Li, Y.Y.; Tan, G.L.; Lan, P.X.; Zhong, L.; Liu, Y.; Li, R.; Li, F. First report of zucchini tigre mosaic virus infecting several cucurbit plants in China. Plant Dis. 2016, 100, 1253. [Google Scholar] [CrossRef]
- Nong, Y. Identification of Viruses Infecting Cucurbit Crops in Guangdong Province and Analysis of Important Virus Characteristics; South China Agricultural University: Guangzhou, China, 2020; pp. 25–26. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayers, E.W.; Cavanaugh, M.; Clark, K.; Ostell, J.; Pruitt, K.D.; Karsch-Mizrachi, I. GenBank. Nucleic Acids Res. 2020, 48, D84–D86. [Google Scholar] [PubMed] [Green Version]
- Koressaar, T.; Lepamets, M.; Kaplinski, L.; Raime, K.; Andreson, R.; Remm, M. Primer3_masker: Integrating masking of template sequence with primer design software. Bioinformatics 2018, 34, 1937–1938. [Google Scholar] [CrossRef] [Green Version]
- Hallgren, J.; Tsirigos, K.D.; Pedersen, M.D.; Armenteros, J.J.A.; Marcatili, P.; Nielsen, H.; Krogh, A.; Winther, O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. BioRxiv 2022. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Varsani, A.; Roumagnac, P.; Botha, G.; Maslamoney, S.; Schwab, T.; Kelz, Z.; Kumar, V.; Murrell, B. RDP5: A computer program for analyzing recombination in, and removing signals of recombination from, nucleotide sequence datasets. Virus Evol. 2021, 7, veaa087. [Google Scholar] [CrossRef]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Virol. 2020, 101, 364–365. [Google Scholar] [CrossRef]
- Peng, C.W.; Peremyslov, V.V.; Mushegian, A.R.; Dawson, W.O.; Dolja, V.V. Functional specialization and evolution of leader proteinases in the family Closteroviridae. J. Virol. 2001, 75, 12153–12160. [Google Scholar] [CrossRef] [Green Version]
- Wintermantel, W.M.; Wisler, G.C.; Anchieta, A.G.; Liu, H.Y.; Karasev, A.V.; Tzanetakis, I.E. The complete nucleotide sequence and genome organization of tomato chlorosis virus. Arch. Virol. 2005, 150, 2287–2298. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Available online: https://ictv.global/report/chapter/closteroviridae/closteroviridae/crinivirus (accessed on 16 April 2023).
- Rubio, L.; Guerri, J.; Moreno, P. Genetic variability and evolutionary dynamics of viruses of the family Closteroviridae. Front. Microbiol. 2013, 4, 151. [Google Scholar] [CrossRef] [Green Version]
- International Committee on Taxonomy of Viruses. Available online: https://ictv.global/report/chapter/endornaviridae/endornaviridae/alphaendornavirus (accessed on 16 April 2023).
- Chen, T.; Zhang, H.Y.; Liu, Y.; Liu, Y.X.; Huang, L.Q. EVenn: Easy to create repeatable and editable Venn diagrams and Venn networks online. J. Genet. Genom. 2021, 48, 863–866. [Google Scholar] [CrossRef]
- Hassan, A.A.; Duffus, J.E. A review of a yellowing and stunting disorder of cucurbits in the United Arab Emirates. Emir. J. Food Agric. 1991, 2, 1–16. [Google Scholar] [CrossRef]
- Coutts, R.H.A.; Livieratos, I.C. Nucleotide sequence and genome organization of cucurbit yellow stunting disorder virus RNA1. Arch. Virol. 2003, 148, 2055–2062. [Google Scholar] [CrossRef]
- Jailani, A.A.K.; Iriarte, F.; Hochmuth, B.; Willis, S.M.; Warren, M.; Dey, K.; Velez-Climentet, M.; McVay, J.; Bag, S.; Paret, M.L. First report of cucurbit chlorotic yellows virus affecting watermelon in USA. Plant Dis. 2022, 106, 774. [Google Scholar] [CrossRef]
- Mondal, S.; Hladky, L.J.; Wintermantel, W.M. Differential seasonal prevalence of yellowing viruses infecting melon crops in southern California and Arizona determined by multiplex RT-PCR and RT-qPCR. Plant Dis. 2023; in press. [Google Scholar] [CrossRef] [PubMed]
- Rubio, L.; Abou-Jawdah, Y.; Lin, H.X.; Falk, B.W. Geographically distant isolates of the crinivirus cucurbit yellow stunting disorder virus show very low genetic diversity in the coat protein gene. J. Gen. Virol. 2001, 82, 929–933. [Google Scholar] [CrossRef] [PubMed]
- Marco, C.F.; Aranda, M.A. Genetic diversity of a natural population of cucurbit yellow stunting disorder virus. J. Gen. Virol. 2005, 86, 815–822. [Google Scholar] [CrossRef]
- Rubio, L.; Soong, J.; Kao, J.; Falk, B.W. Geographic distribution and molecular variation of isolates of three whitefly-borne closteroviruses of cucurbits: Lettuce infectious yellows virus, cucurbit yellow stunting disorder virus, and beet pseudo-yellows virus. Phytopathology 1999, 89, 707–711. [Google Scholar] [CrossRef] [Green Version]
- Boubourakas, I.N.; Avgelis, A.D.; Kyriakopoulou, P.E.; Katis, N.I. Occurrence of yellowing viruses (Beet pseudo-yellows virus, cucurbit yellow stunting disorder virus and cucurbit aphid-borne yellows virus) affecting cucurbits in Greece. Plant Pathol. 2006, 55, 276–283. [Google Scholar] [CrossRef]
- Krishnan, N.; Kumari, S.; Pandey, S.; Ram, D.; Behera, T.K.; Gandhi, K. Occurrence of cucurbit yellow stunting disorder virus causing yellowing disease of cucurbits in India. Crop Protect. 2022, 158, 106013. [Google Scholar] [CrossRef]
- Fuchs, M. Closteroviruses (Closteroviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: New York, NY, USA, 2021; pp. 336–347. ISBN 9780128145166. [Google Scholar]
- Grill, L.K.; Garger, S.J. Identification and characterization of double-stranded RNA associated with cytoplasmic male sterility in Vicia faba. Proc. Natl. Acad. Sci. USA 1981, 78, 7043–7046. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, T. Endornaviruses: Persistent dsRNA viruses with symbiotic properties in diverse eukaryotes. Virus Genes 2019, 55, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Fukuhara, T. Endornaviruses (Endornaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: New York, NY, USA, 2021; pp. 388–395. ISBN 9780128145166. [Google Scholar]
- Sabanadzovic, S.; Wintermantel, W.M.; Valverde, R.A.; McCreight, J.D.; Aboughanem-Sabanadzovic, N. Cucumis melo endornavirus: Genome organization, host range and co-divergence with the host. Virus Res. 2016, 214, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khankhum, S.; Valverde, R.A.; Pastor-Corrales, M.A.; Osorno, J.M.; Sabanadzovic, S. Two endornaviruses show differential infection patterns between gene pools of Phaseolus vulgaris. Arch. Virol. 2015, 160, 1131–1137. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarria, F.; Shen, G.; Roe, B. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef]
- Kato, K.; Hanada, K.; Kameya-Iwaki, M. Transmissions mode, host range and electron microscopy of a pathogen causing a new disease of melon (Cucumis melo) in Japan. Ann. Phytopathol. Soc. Jpn. 1999, 65, 624–627. [Google Scholar] [CrossRef]
- Quito-Avila, D.F.; Peralta, E.L.; Martin, R.R.; Ibarra, M.A.; Alvarez, R.A.; Mendoza, A.; Insuasti, M.; Ochoa, J. Detection and occurrence of melon yellow spot virus in Ecuador: An emerging threat to cucurbit production in the region. Eur. J. Plant Pathol. 2014, 140, 193–197. [Google Scholar] [CrossRef]
- Che, H.Y.; Cao, X.R.; He, Y.H.; Luo, D.Q. Distribution and identification of watermelon viruses in Hainan Island. Acta Phytopathol. Sin. 2020, 50, 632–636. [Google Scholar]
- Orfanidou, C.G.; Baltzi, A.; Dimou, N.A.; Katis, N.I.; Maliogka, V.I. Cucurbit chlorotic yellows virus: Insights into its natural host range, genetic variability, and transmission parameters. Plant Dis. 2017, 101, 2053–2058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamian, P.; Sobh, H.; Seblani, R.; Abou-Jawdah, Y. Co-infection of two criniviruses and a begomovirus enhances the disease severity in cucumber. Eur. J. Plant Pathol. 2015, 142, 521–530. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Genus | Virus (Acc. No. of Protein) | No. of Contigs | Length Range (nt) | Identity Range (%) |
|---|---|---|---|---|---|
| Closteroviridae | Crinivirus | cucurbit yellow stunting disorder virus (NP_851566, NP_851572, NP_851576 and NP_851578) | 5 | 1328–8886 | 66.1–83.9 |
| Closteroviridae | Crinivirus | cucurbit chlorotic yellows virus (QFI57123, QFI57127, QFI57128, QFI57130 and YP_006522423) | 7 | 904–8611 | 99.6–100 |
| Tospoviridae | Orthotospovirus | watermelon silver mottle virus (QKX94930, NP_620752, NP_620767, NP_620770 and NP_620771) | 6 | 465–7584 | 94.4–98.5 |
| Tospoviridae | Orthotospovirus | melon yellow spot virus (YP_717933, YP_717935, QIG37604, QIG37605 and QIG37606) | 8 | 251–8902 | 94.2–100 |
| Potyviridae | Potyvirus | papaya ringspot virus (NP_056758 and BBK60930) | 3 | 270–10,320 | 91.2–95.5 |
| Endornaviridae | Alphaendornavirus | cucumis melo endornavirus (QIZ03212) | 2 | 14,990–15,020 | 64–64.2 |
| Isolate | Host | Symptoms | Origin | Accession Numbers (RNA1, RNA2) |
|---|---|---|---|---|
| Chieh-qua-CM | Benincasa hispida Cogn. var. chieh-qua How | Chlorotic, mottling | Chengmai, Hainan | OP976048, OP976049 |
| Watermelon-LS | Cucurbita moschata | Chlorotic, mosaic | Lingshui, Hainan | OP976050, OP976051 |
| Pumpkin-DF | Citrullus lanatus | Chlorotic, mosaic | Dongfang, Hainan | OP976052, OP976053 |
| Cucumber-SY | Cucumis sativus | Chlorotic, mosaic | Sanyan, Hainan | OP976054, OP976055 |
| Location | Percentage of Positive Samples (No. of Virus Infected Samples/Total No. of Plants Analyzed) | |||||||
|---|---|---|---|---|---|---|---|---|
| MYSV | CCYV | CqEV | CuCV | WSMoV | PRSV | CMV | ZYMV | |
| Chengmai | 36.11 (13/36) | 33.33 (12/36) | 22.22 (8/36) | 33.33 (12/36) | 5.56 (2/36) | 5.56 (2/36) | 0 (0/36) | 2.78 (1/36) |
| Wenchang | 30.43 (7/23) | 69.57 (16/23) | 21.74 (5/23) | 13.04 (3/23) | 0 (0/23) | 0 (0/23) | 8.7 (2/23) | 0 (0/23) |
| Ding’an | 100 (14/14) | 57.14 (8/14) | 92.86 (13/14) | 28.57 (4/14) | 0 (0/14) | 7.14 (1/14) | 28.57 (4/14) | 0 (0/14) |
| Lingshui | 100 (9/9) | 61.54 (8/13) | 33.33(3/9) | 100 (9/9) | 88.89 (8/9) | 33.33 (3/9) | 0 (0/14) | 11.11 (1/9) |
| Sanya | 100 (18/18) | 66.67 (12/18) | 16.67 (3/18) | 50 (9/18) | 0 (0/18) | 11.11(2/18) | 11.11(2/18) | 11.11 (2/18) |
| Dongfang | 100 (13/13) | 23.53 (4/17) | 100(13/13) | 0 (0/13) | 0 (0/13) | 7.69 (1/13) | 0 (0/13) | 0 (0/13) |
| Danzhou | 72.73 (16/22) | 68.18 (15/22) | 13.64 (3/22) | 0 (0/22) | 0 (0/22) | 0 (0/22) | 13.64 (3/22) | 22.73 (5/22) |
| Total | 66.67 (90/135) | 55.56 (75/135) | 35.56 (48/135) | 27.41(37/135) | 7.41 (10/135) | 6.67 (9/135) | 8.15(11/135) | 6.67 (9/135) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Che, H.; Ma, Y.; Lin, Y.; Feng, T.; Luo, D.; Long, H. Virome Profiling, New Virus Identification and the Prevalence and Distribution of Viruses Infecting Chieh-Qua (Benincasa hispida Cogn. var. chieh-qua How) in China. Viruses 2023, 15, 1396. https://doi.org/10.3390/v15061396
Che H, Ma Y, Lin Y, Feng T, Luo D, Long H. Virome Profiling, New Virus Identification and the Prevalence and Distribution of Viruses Infecting Chieh-Qua (Benincasa hispida Cogn. var. chieh-qua How) in China. Viruses. 2023; 15(6):1396. https://doi.org/10.3390/v15061396
Chicago/Turabian StyleChe, Haiyan, Yuxin Ma, Yating Lin, Tuizi Feng, Daquan Luo, and Haibo Long. 2023. "Virome Profiling, New Virus Identification and the Prevalence and Distribution of Viruses Infecting Chieh-Qua (Benincasa hispida Cogn. var. chieh-qua How) in China" Viruses 15, no. 6: 1396. https://doi.org/10.3390/v15061396