

Genomic Characterization of a Halovirus Representing a Novel Siphoviral Cluster

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods Sterile
2.1. Isolation of Host H. titanicae H5 and Phage YPHTV-1
2.2. Transmission Electron Microscopy
2.3. Host Range
2.4. One-Step Growth Curve
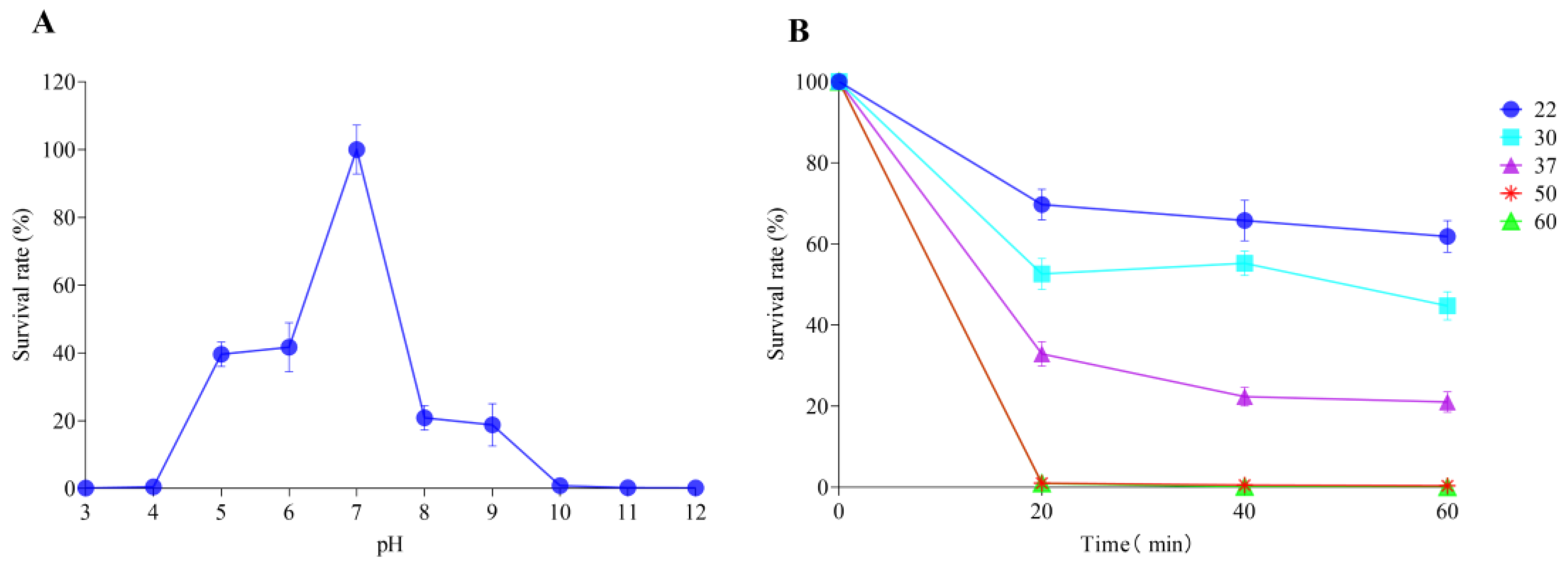
2.5. pH and Thermal Stability
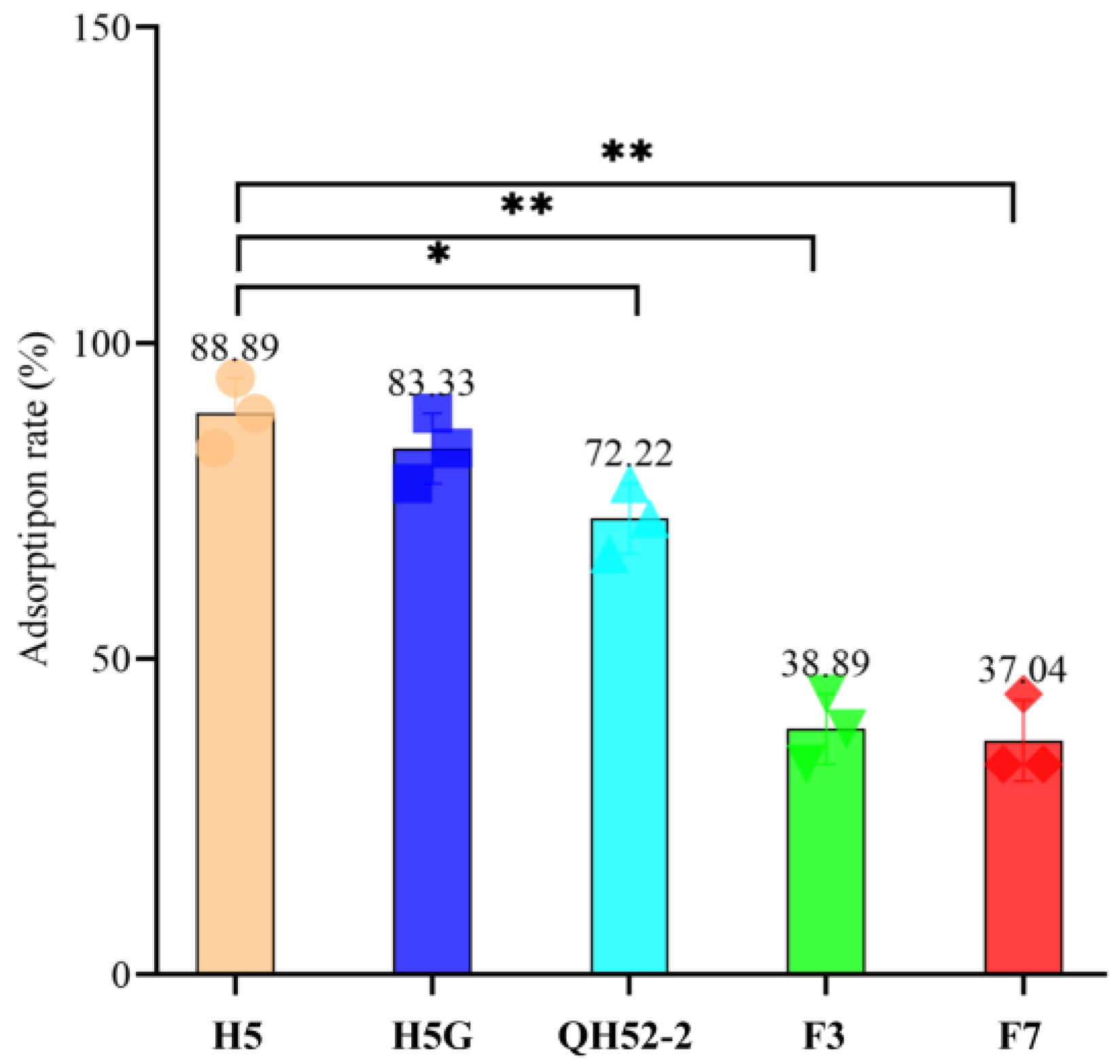
2.6. Phage Adsorption Rate to Different Cells
2.7. Preparation of High-Titer Phage Lysate
2.8. Extraction, Sequencing, and Bioinformatic Analysis of Phage DNA
3. Results
3.1. Biological Characteristics of YPHTV-1
3.2. Genomic Property of Phage YPHTV-1
3.3. DNA Replication and Virion Assembly
3.4. Auxiliary Metabolic Genes
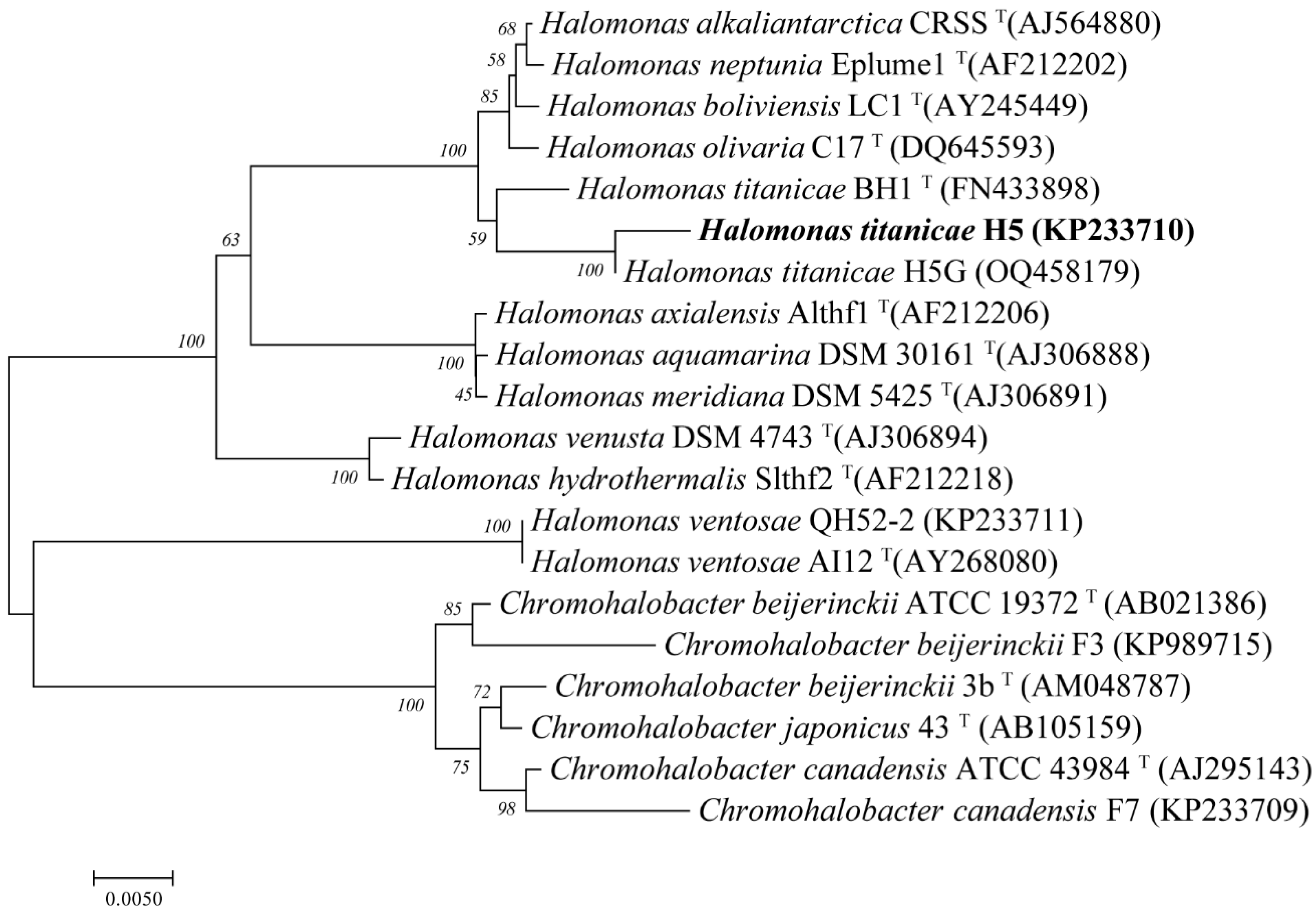
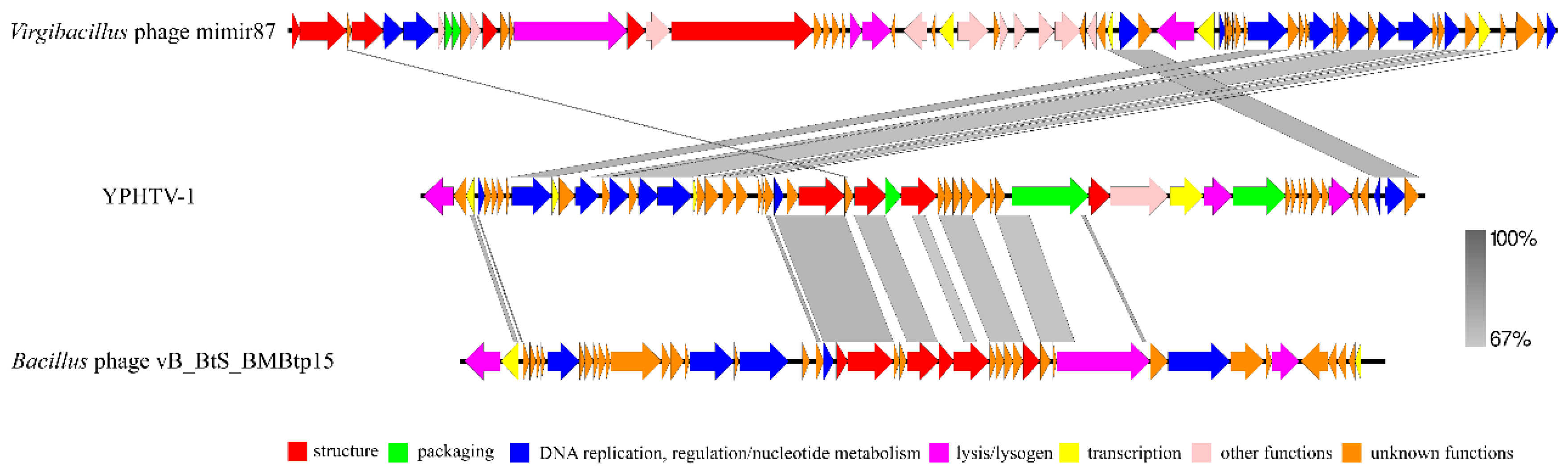
3.5. Phage YPHTV-1 Represents a New Cluster
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.-B.; Rodriguez-Brito, B.; Pasic, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Narasingarao, P.; Podell, S.; Ugalde, J.A.; Brochier-Armanet, C.; Emerson, J.B.; Brocks, J.J.; Heidelberg, K.B.; Banfield, J.F.; Allen, E.E. De novo metagenomic assembly reveals abundant novel major lineage of Archaea in hypersaline microbial communities. ISME J. 2012, 6, 81–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Haloviruses of archaea, bacteria, and eukaryotes. Curr. Opin Microbiol. 2015, 25, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, N.S.; Bamford, D.H.; Oksanen, H.M. Virus-host interplay in high salt environments. Environ. Microbiol. Rep. 2016, 8, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Boujelben, I.; Yarza, P.; Almansa, C.; Villamor, J.; Maalej, S.; Antón, J.; Santos, F. Virioplankton Community Structure in Tunisian Solar Salterns. Appl. Environ. Microbiol. 2012, 78, 7429–7437. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Heredia, I.; Martin-Cuadrado, A.-B.; Mojica, F.J.M.; Santos, F.; Mira, A.; Antón, J.; Rodriguez-Valera, F. Reconstructing Viral Genomes from the Environment Using Fosmid Clones: The Case of Haloviruses. PLoS ONE 2012, 7, e33802. [Google Scholar] [CrossRef] [Green Version]
- Witte, A.; Baranyi, U.; Klein, R.; Sulzner, M.; Luo, C.; Wanner, G.; Kru¨ger, D.H.; Lubitz, W. Characterization of Natronobacterium magadii phage φCh1, a unique archaeal phage containing DNA and RNA. Mol. Microbiol. 1997, 23, 603–616. [Google Scholar] [CrossRef]
- Pagaling, E.; Haigh, R.D.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Sequence analysis of an Archaeal virus isolated from a hypersaline lake in Inner Mongolia, China. BMC Genom. 2007, 8, 410. [Google Scholar] [CrossRef] [Green Version]
- Pietilä, M.K.; Laurinmäki, P.; Russell, D.A.; Ko, C.-C.; Jacobs-Sera, D.; Hendrix, R.W.; Bamford, D.H.; Butcher, S.J. Structure of the archaeal head-tailed virus HSTV-1 completes the HK97 fold story. PNAS 2013, 110, 10604–10609. [Google Scholar] [CrossRef] [Green Version]
- Pietilä, M.K.; Roine, E.; Paulin, L.; Kalkkinen, N.; Bamford, D.H. An ssDNA virus infecting archaea: A new lineage of viruses with a membrane envelope. Mol. Microbiol. 2009, 72, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Dyall-Smith, M.; Tang, S.-L.; Bath, C. Haloarchaeal viruses: How diverse are they? Res. Microbiol. 2003, 154, 309–313. [Google Scholar] [CrossRef]
- Pietilä, M.K.; Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Modified coat protein forms the flexible spindle-shaped virion of haloarchaeal virus His1. Environ. Microbiol. 2013, 15, 1674–1686. [Google Scholar] [CrossRef] [PubMed]
- Calvo, C.; García de la Paz, A.; Pérez-Martínez, A.; Ramos-Cormenzana, A. Isolation of phages HM5 and HM15 from hypersaline soil. Toxicol. Environ. Chem. 1991, 34, 29–37. [Google Scholar] [CrossRef]
- Kauri, T.; Ackermann, H.-W.; Goel, U.; Kushner, D.J. A bacteriophage of a moderately halophilic bacterium. Arch. Microbiol. 1991, 156, 435–438. [Google Scholar] [CrossRef]
- Calvo, C.; Paz, A.M.G.D.L.; Caba, F.M.C.M.A. Behaviour of two D. halophila bacteriophages with respect to salt concentrations and other environmental factors. Toxicol. Environ. Chem. 1994, 43, 85–93. [Google Scholar] [CrossRef]
- Villamor, J.; Ramos-Barbero, M.D.; González-Torres, P.; Gabaldón, T.; Rosselló-Móra, R.; Meseguer, I.; Martínez-García, M.; Santos, F.; Antón, J. Characterization of ecologically diverse viruses infecting co-occurring strains of cosmopolitan hyperhalophilic Bacteroidetes. ISME J. 2018, 12, 424–437. [Google Scholar] [CrossRef]
- Wang, C.-X.; Li, X. JMT-1: A novel, spherical lytic halotolerant phage isolated from Yuncheng saline lake. Braz. J. Microbiol. 2018, 49, 262–268. [Google Scholar] [CrossRef]
- Wang, C.-X.; Zhao, A.-H.; Yu, H.-Y.; Wang, L.-L.; Li, X. Isolation and Characterization of a Novel Lytic Halotolerant Phage from Yuncheng Saline Lake. Indian J. Microbiol. 2022, 62, 249–256. [Google Scholar] [CrossRef]
- Mobberley, J.M.; Authement, R.N.; Segall, A.M.; Paul, J.H. The Temperate Marine Phage ΦHAP-1 of Halomonas aquamarina Possesses a Linear Plasmid-Like Prophage Genome. J. Virol. 2008, 82, 6618–6630. [Google Scholar] [CrossRef] [Green Version]
- Olonade, I.; van Zyl, L.J.; Trindade, M. Genomic characterization of a prophage, Smhb1, that infects Salinivibrio kushneri BNH isolated from a namib desert saline spring. Microorganisms 2021, 9, 2043. [Google Scholar] [CrossRef] [PubMed]
- Rodela, M.L.; Sabet, S.; Peterson, A.; Dillon, J.G. Broad environmental tolerance for a Salicola host-phage pair isolated from the Cargill Solar Saltworks, Newark, CA, USA. Microorganisms 2019, 7, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aalto, A.P.; Bitto, D.; Ravantti, J.J.; Bamford, D.H.; Huiskonen, J.T.; Oksanen, H.M. Snapshot of virus evolution in hypersaline environments from the characterization of a membrane-containing Salisaeta icosahedral phage 1. Proc. Natl. Acad. Sci. USA 2012, 109, 7079–7084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, P.S.; Domek, M.J.; Sanz-García, E.; Makaju, A.; Taylor, R.M.; Hoggan, R.; Culumber, M.D.; Oberg, C.J.; Breakwell, D.P.; Prince, J.T.; et al. Sequence and Structural Characterization of Great Salt Lake Bacteriophage CW02, a Member of the T7-Like Supergroup. J. Virol. 2012, 86, 7907–7917. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Zhao, Q.; Li, Z.; Wang, Y.; Zhang, S.; Lai, Y.; Xiao, W.; Cui, X. Complete genome sequence of Halomonas ventosae virulent halovirus QHHSV-1. Arch. Virol. 2017, 162, 3215–3219. [Google Scholar] [CrossRef]
- Zrelovs, N.; Cernooka, E.; Dislers, A.; Kazaks, A. Isolation and characterization of the novel Virgibacillus-infecting bacteriophage Mimir87. Arch. Virol. 2020, 165, 737–741. [Google Scholar] [CrossRef]
- Yi, H.; Fu, C.; Diao, K.; Li, Z.; Cui, X.; Xiao, W. Characterization and genomic analysis of a novel halovirus infecting Chromohalobacter beijerinckii. Front. Microbiol. 2022, 13, 1041471. [Google Scholar] [CrossRef]
- Boughalmi, M.; Saadi, H.; Pagnier, I.; Colson, P.; Fournous, G.; Raoult, D.; La Scola, B. High-throughput isolation of giant viruses of the Mimiviridae and Marseilleviridae families in the Tunisian environment. Environ. Microbiol. 2013, 15, 2000–2007. [Google Scholar] [CrossRef]
- Whitman, W.B.; Rainey, F.; Kämpfer, P.; Trujillo, M.; Chun, J.; DeVos, P. Halomonas. In Bergey’s Manual of Systematics of Archaea and Bacteria; Wiley: Hoboken, NJ, USA, 2015; pp. 1–19. [Google Scholar]
- Chen, Z.; Wan, C. Non-sterile fermentations for the economical biochemical conversion of renewable feedstocks. Biotechnol. Lett. 2017, 39, 1765–1777. [Google Scholar] [CrossRef]
- Tan, D.; Xue, Y.-S.; Aibaidula, G.; Chen, G.-Q. Unsterile and continuous production of polyhydroxybutyrate by Halomonas TD01. Bioresour. Technol. 2011, 102, 8130–8136. [Google Scholar] [CrossRef]
- Seaman, P.F.; Day, M.J. Isolation and characterization of a bacteriophage with an unusually large genome from the Great Salt Plains National Wildlife Refuge, Oklahoma, USA. FEMS Microbiol. Ecol. 2007, 60, 1–13. [Google Scholar] [CrossRef]
- Calvo, C.; De La Paz, A.G.; Béjar, V.; Quesada, E.; Ramos-Cormenzana, A. Isolation and characterization of phage F9-11 from a lysogenicDeleya halophila strain. Curr. Microbiol. 1988, 17, 49–53. [Google Scholar] [CrossRef]
- Parro, V.; de Diego-Castilla, G.; Moreno-Paz, M.; Blanco, Y.; Cruz-Gil, P.; Rodríguez-Manfredi, J.A.; Fernández-Remolar, D.; Gómez, F.; Gómez, M.J.; Rivas, L.A.; et al. A Microbial Oasis in the Hypersaline Atacama Subsurface Discovered by a Life Detector Chip: Implications for the Search for Life on Mars. Astrobiology 2011, 11, 969–996. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, Y.-X.; Liu, J.-H.; Wang, Z.-G.; Zhang, X.-X.; Ji, K.-Y.; Lai, Y.-H.; Wen, M.-L.; Cui, X.-L. Roseivivax sediminis sp. nov., a moderately halophilic bacterium isolated from salt mine sediment. Int. J. Syst. Evol. Microbiol. 2012, 62 Pt 8, 1890–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.-Q.; Zhao, Q.; Li, Z.-Y.; Wang, Y.-X.; Zhang, S.-Y.; Lai, Y.-H.; Xiao, W.; Cui, X.-L. A novel Halomonas ventosae-specific virulent halovirus isolated from the Qiaohou salt mine in Yunnan, Southwest China. Extremophiles 2015, 20, 101–110. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Pirovano, W. Toward almost closed genomes with GapFiller. Genome Biol. 2012, 13, R56. [Google Scholar] [CrossRef] [Green Version]
- Besemer, J.; Alexandre, L.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrascosa, J.L.; Camacho, A.; Moreno, F.; Jimenez, F.; Mellado, R.P.; Viñuela, E.; Salas, M. Bacillus subtilis Phage phi 29 Characterization of Gene Products and Functions. JBIC J. Biol. Inorg. Chem. 1976, 66, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Li, M.; Zhao, C.; Yang, K.; Li, K.; Peng, M.; Yang, Z.; Liu, F.; Liu, Y.; Bai, R.; et al. Concentrations of toxic metals and ecological risk assessment for sediments of major freshwater lakes in China. J. Geochem. Explor. 2015, 157, 15–26. [Google Scholar] [CrossRef]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.L. HNH proteins are a widespread component of phage DNA packaging machines. Proc. Natl. Acad. Sci. USA 2014, 111, 6022–6027. [Google Scholar] [CrossRef] [Green Version]
- Quiles-Puchalt, N.; Carpena, N.; Alonso, J.C.; Novick, R.P.; Marina, A.; Penadés, J.R. Staphylococcal pathogenicity island DNA packaging system involving cos -site packaging and phage-encoded HNH endonucleases. Proc. Natl. Acad. Sci. USA 2014, 111, 6016–6021. [Google Scholar] [CrossRef] [Green Version]
- Duda, R.L.; Martincic, K.; Hendrix, R.W. Genetic basis of bacteriophage HK97 prohead assembly. J. Mol. Biol. 1995, 247, 636–647. [Google Scholar] [CrossRef]
- Bose, B.; Auchtung, J.M.; Lee, C.A.; Grossman, A.D. A conserved anti-repressor controls horizontal gene transfer by proteolysis. Mol. Microbiol. 2008, 70, 570–582. [Google Scholar] [CrossRef] [Green Version]
- Ludanyi, M.; Blanchard, L.; Dulermo, R.; Brandelet, G.; Bellanger, L.; Pignol, D.; Lemaire, D.; de Groot, A. Radiation response in Deinococcus deserti: IrrE is a metalloprotease that cleaves repressor protein DdrO. Mol. Microbiol. 2014, 94, 434–449. [Google Scholar] [CrossRef]
- Vujicic-Zagar, A.; Dulermo, R.; Le Gorrec, M.; Vannier, F.; Servant, P.; Sommer, S.; de Groot, A.; Serre, L. Crystal structure of the IrrE protein, a central regulator of DNA damage repair in deinococcaceae. J. Mol. Biol. 2009, 386, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Vera, M.; Pagliai, F.; Guiliani, N.; Jerez, C.A. The Chemolithoautotroph Acidithiobacillus ferrooxidans Can Survive under Phosphate-Limiting Conditions by Expressing a C-P Lyase Operon that Allows It to Grow on Phosphonates. Appl. Environ. Microbiol. 2008, 74, 1829–1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clokie, M.R.J.; Mann, N.H. Marine cyanophages and light. Environ. Microbiol. 2006, 8, 2074–2082. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, Z.; Chen, J.; Ernst, R.K.; Wang, X. Influence of Lipid A Acylation Pattern on Membrane Permeability and Innate Immune Stimulation. Mar. Drugs 2013, 11, 3197–3208. [Google Scholar] [CrossRef] [Green Version]
- Vreeland, R.H.; Rosenzweig, W.D.; Powers, D.W. Isolation of a 250 million-year-old halotolerant bacterium from a primary salt crystal. Nature 2000, 407, 897–900. [Google Scholar] [CrossRef]
- Fu, Y.; Deng, S.; Liang, L.; Wu, Y.; Gao, M. Complete genome sequence of the novel phage vB_BthS-HD29phi infecting Bacillus thuringiensis. Arch. Virol. 2019, 164, 3089–3093. [Google Scholar] [CrossRef]
- Gillis, A.; Mahillon, J. Phages Preying on Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis: Past, Present and Future. Viruses 2014, 6, 2623–2672. [Google Scholar] [CrossRef] [Green Version]
- Seed, K.D. Battling Phages: How Bacteria Defend against Viral Attack. PLOS Pathog. 2015, 11, e1004847. [Google Scholar] [CrossRef] [Green Version]
- Tock, M.R.; Dryden, D.T. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 2005, 8, 466–472. [Google Scholar] [CrossRef]
- Bragg, J.G.; Chisholm, S.W. Modeling the Fitness Consequences of a Cyanophage-Encoded Photosynthesis Gene. PLoS ONE 2008, 3, e3550. [Google Scholar] [CrossRef] [Green Version]
- Mann, N.H.; Cook, A.; Millard, A.; Bailey, S.; Clokie, M. Bacterial photosynthesis genes in a virus. Nature 2003, 424, 741. [Google Scholar] [CrossRef]
- Breitbart, M.; Thompson, L.; Suttle, C.; Sullivan, M. Exploring the Vast Diversity of Marine Viruses. Oceanography 2007, 20, 135–139. [Google Scholar] [CrossRef]
- Thompson, L.R.; Zeng, Q.; Kelly, L.; Huang, K.H.; Singer, A.U.; Stubbe, J.; Chisholm, S.W. Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. USA 2011, 108, E757–E764. [Google Scholar] [CrossRef] [Green Version]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Burgos, M.; García-Romero, I.; Jung, J.; Valvano, M.A.; Søgaard-Andersen, L. Identification of the lipopolysaccharide O-antigen biosynthesis priming enzyme and the O-antigen ligase in Myxococcus xanthus: Critical role of LPS O-antigen in motility and development. Mol. Microbiol. 2019, 112, 1178–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, M.; Marianovsky, I.; Glaser, G. MazG–A regulator of programmed cell death in Escherichia coli. Mol. Microbiol. 2006, 59, 590–601. [Google Scholar] [CrossRef]
- Duhaime, M.B.; Solonenko, N.; Roux, S.; Verberkmoes, N.C.; Wichels, A.; Sullivan, M.B. Comparative Omics and Trait Analyses of Marine Pseudoalteromonas Phages Advance the Phage OTU Concept. Front. Microbiol. 2017, 8, 1241. [Google Scholar] [CrossRef]
- Mikoulinskaia, G.V.; Odinokova, I.V.; Zimin, A.A.; Lysanskaya, V.Y.; Feofanov, S.A.; Stepnaya, O.A. Identification and characterization of the metal ion-dependent l-alanoyl-d-glutamate peptidase encoded by bacteriophage T5. FEBS J. 2009, 276, 7329–7342. [Google Scholar] [CrossRef]
- Antonova, N.P.; Vasina, D.V.; Lendel, A.M.; Usachev, E.V.; Makarov, V.V.; Gintsburg, A.L.; Tkachuk, A.P.; Gushchin, V.A. Broad bactericidal activity of the Myoviridae bacteriophage lysins LysAm24, LysECD7, and LysSi3 against Gram-negative ESKAPE pathogens. Viruses 2019, 11, 284. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diao, K.; Li, G.; Sun, X.; Yi, H.; Zhang, S.; Xiao, W. Genomic Characterization of a Halovirus Representing a Novel Siphoviral Cluster. Viruses 2023, 15, 1392. https://doi.org/10.3390/v15061392
Diao K, Li G, Sun X, Yi H, Zhang S, Xiao W. Genomic Characterization of a Halovirus Representing a Novel Siphoviral Cluster. Viruses. 2023; 15(6):1392. https://doi.org/10.3390/v15061392
Chicago/Turabian StyleDiao, Kaixin, Guohui Li, Xueqin Sun, Hao Yi, Shiying Zhang, and Wei Xiao. 2023. "Genomic Characterization of a Halovirus Representing a Novel Siphoviral Cluster" Viruses 15, no. 6: 1392. https://doi.org/10.3390/v15061392