
The Autonomous Parvovirus Minute Virus of Mice Localizes to Cellular Sites of DNA Damage Using ATR Signaling

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus, Viral Infections
2.2. Cell Synchronization, DNA Damage, and Drug Treatments
2.3. Plasmids and Transfections
2.4. Laser Micro-Irradiation for Immunofluorescence
2.5. Immunofluorescence Assays
2.6. Image Analysis
2.7. Cell Cycle Analysis
2.8. MVMp Genome Replication Analysis
2.9. Salt-Wash Chromatin Immunoprecipitation Combined with Quantitative PCR (swChIP-qPCR)
2.10. Statistical Analysis
2.11. Western Blot Analysis
2.12. Antibodies
3. Results
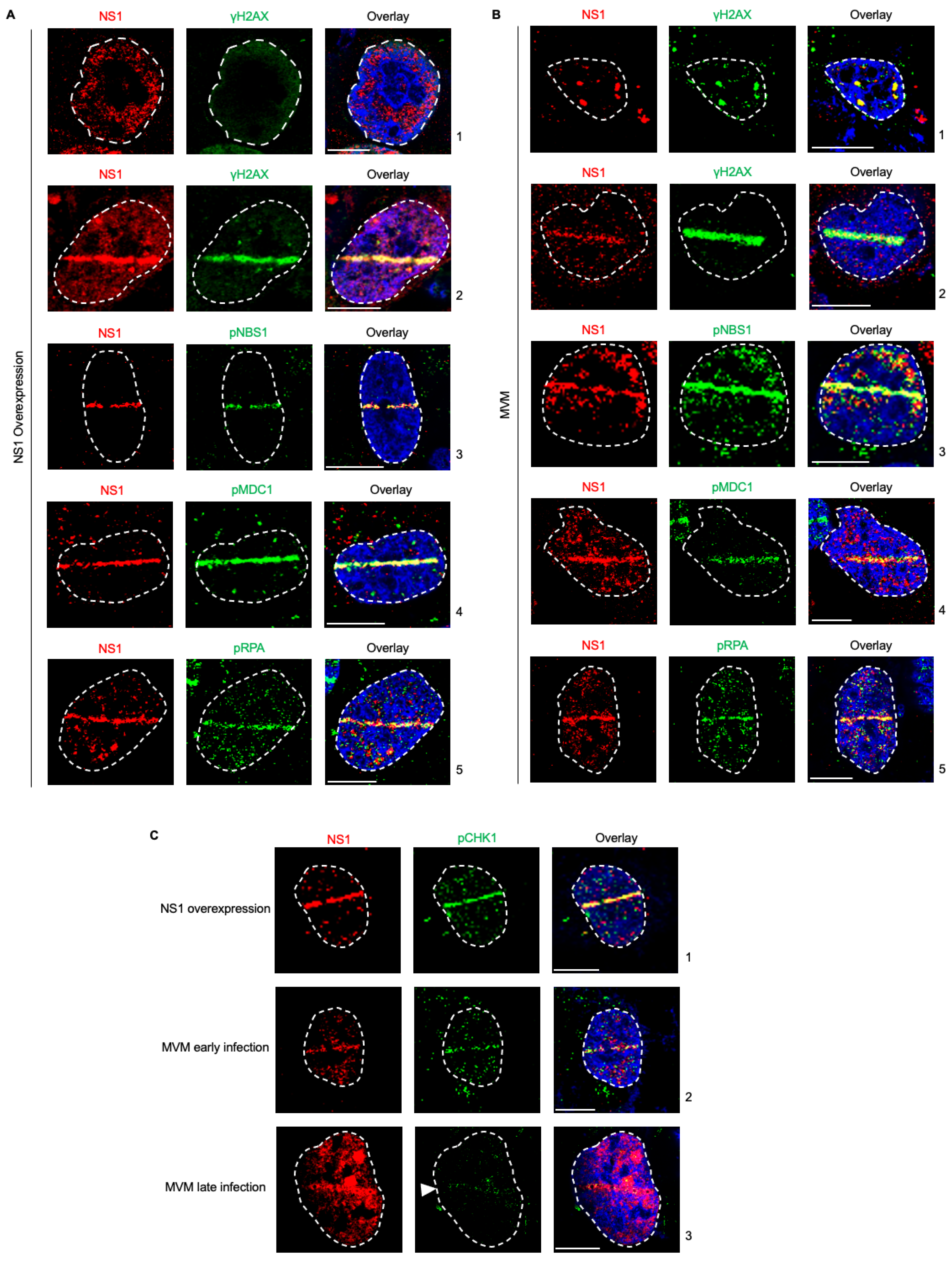
3.1. NS1 Is a Proxy Marker for Cellular DNA Damage during MVM Infection
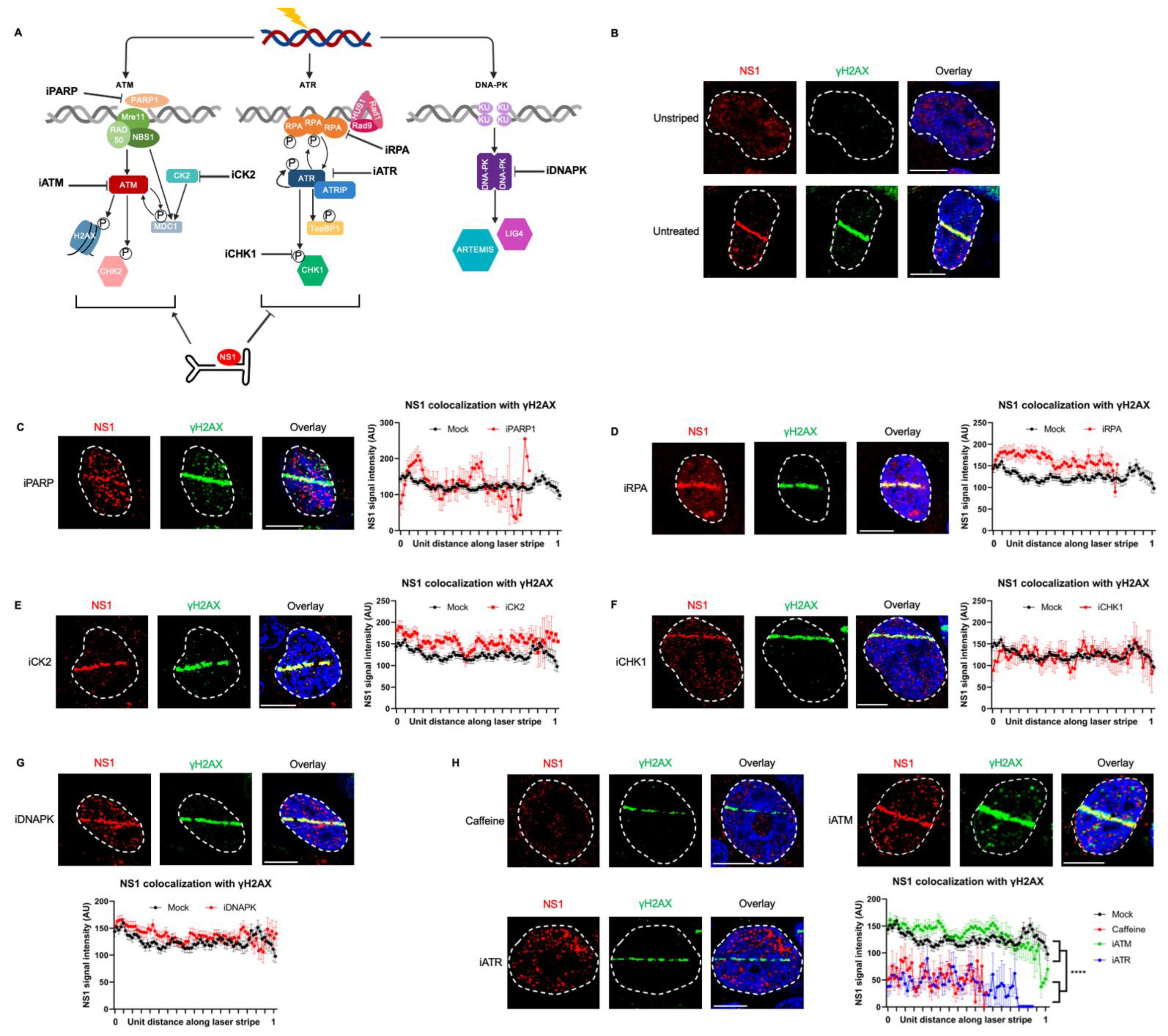
3.2. NS1 Localization to Cellular DDR Sites Is Independent of ATM and DNA-PK Signaling but Depends on ATR Signaling
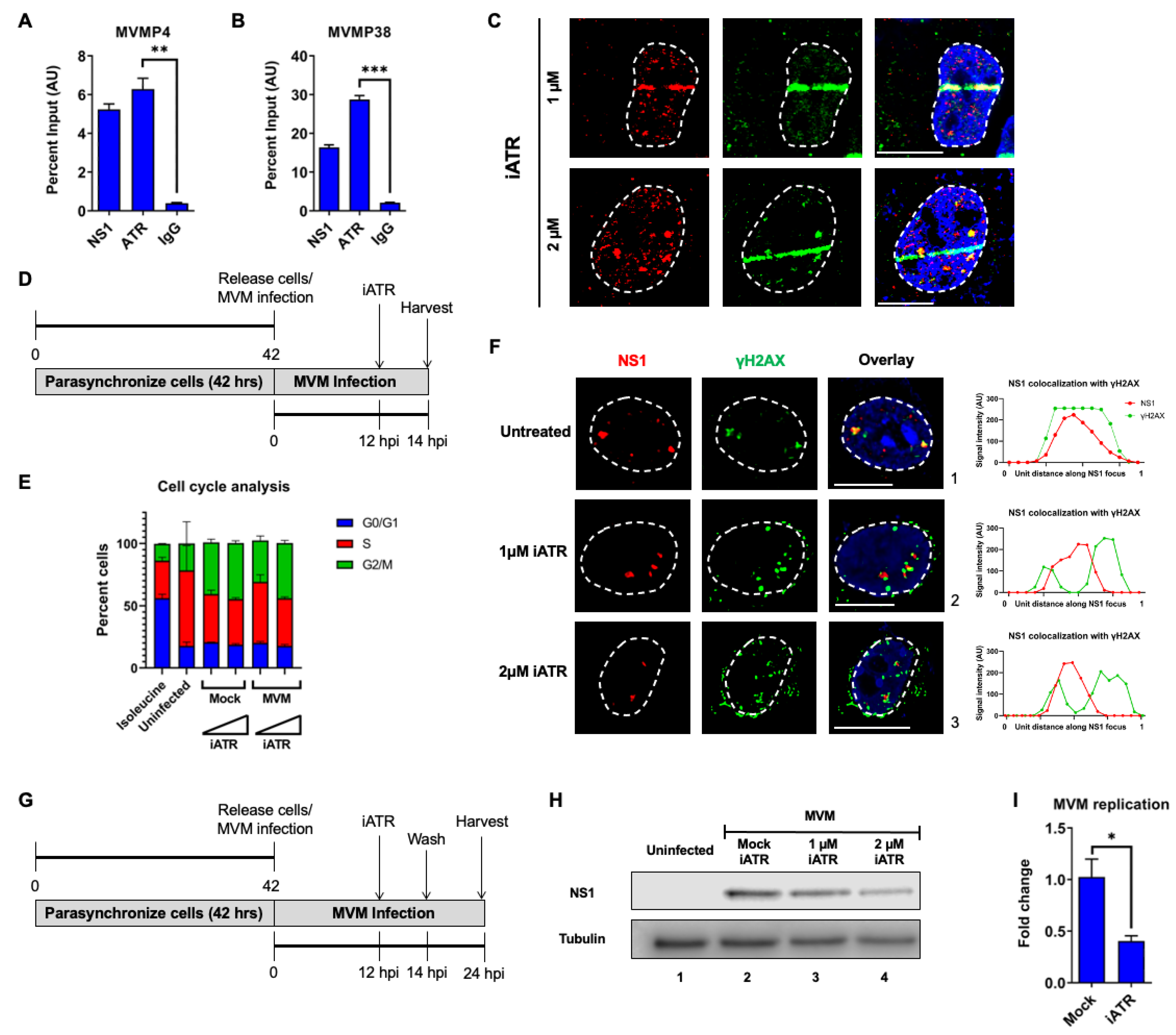
3.3. ATR Associates with the MVM Genome and ATR Signaling Regulates the Early Replication of MVM
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviruses: Small does not mean simple. Annu. Rev. Virol. 2014, 1, 517–537. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. The ns-1 polypeptide of minute virus of mice is covalently attached to the 5’ termini of duplex replicative-form dna and progeny single strands. J. Virol. 1988, 62, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Christensen, J.; Nüesch, J.P.; Tattersall, P. The ns1 polypeptide of the murine parvovirus minute virus of mice binds to dna sequences containing the motif [acca]2-3. J. Virol. 1995, 69, 1652–1660. [Google Scholar] [CrossRef]
- Christensen, J.; Tattersall, P. Parvovirus initiator protein ns1 and rpa coordinate replication fork progression in a reconstituted dna replication system. J. Virol. 2002, 76, 6518–6531. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Gottlieb, R.L.; Tattersall, P. Replication initiator protein ns1 of the parvovirus minute virus of mice binds to modular divergent sites distributed throughout duplex viral dna. J. Virol. 2007, 81, 13015–13027. [Google Scholar] [CrossRef]
- Mouw, M.; Pintel, D.J. Amino acids 16-275 of minute virus of mice ns1 include a domain that specifically binds (acca)2-3-containing dna. Virology 1998, 251, 123–131. [Google Scholar] [CrossRef]
- Naeger, L.K.; Cater, J.; Pintel, D.J. The small nonstructural protein (ns2) of the parvovirus minute virus of mice is required for efficient dna replication and infectious virus production in a cell-type-specific manner. J. Virol. 1990, 64, 6166–6175. [Google Scholar] [CrossRef]
- Bashir, T.; Horlein, R.; Rommelaere, J.; Willwand, K. Cyclin a activates the dna polymerase delta -dependent elongation machinery in vitro: A parvovirus dna replication model. Proc. Natl. Acad. Sci. USA 2000, 97, 5522–5527. [Google Scholar] [CrossRef]
- Bashir, T.; Rommelaere, J.; Cziepluch, C. In vivo accumulation of cyclin a and cellular replication factors in autonomous parvovirus minute virus of mice-associated replication bodies. J. Virol. 2001, 75, 4394–4398. [Google Scholar] [CrossRef]
- Tattersall, P.; Ward, D.C. Rolling hairpin model for replication of parvovirus and linear chromosomal dna. Nature 1976, 263, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Young, P.J.; Jensen, K.T.; Burger, L.R.; Pintel, D.J.; Lorson, C.L. Minute virus of mice ns1 interacts with the smn protein, and they colocalize in novel nuclear bodies induced by parvovirus infection. J. Virol. 2002, 76, 3892–3904. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, Z.; Mihaylov, I.S.; Cotmore, S.F.; Tattersall, P. Recruitment of dna replication and damage response proteins to viral replication centers during infection with ns2 mutants of minute virus of mice (mvm). Virology 2011, 410, 375–384. [Google Scholar] [CrossRef] [PubMed]
- Adeyemi, R.O.; Landry, S.; Davis, M.E.; Weitzman, M.D.; Pintel, D.J. Parvovirus minute virus of mice induces a dna damage response that facilitates viral replication. PLoS Pathog. 2010, 6, e1001141. [Google Scholar] [CrossRef]
- Adeyemi, R.O.; Pintel, D.J. Parvovirus-induced depletion of cyclin b1 prevents mitotic entry of infected cells. PLoS Pathog. 2014, 10, e1003891. [Google Scholar] [CrossRef]
- Fuller, M.S.; Majumder, K.; Pintel, D.J. Minute virus of mice inhibits transcription of the cyclin b1 gene during infection. J. Virol. 2017, 91, e00428-17. [Google Scholar] [CrossRef]
- Pancholi, N.J.; Price, A.M.; Weitzman, M.D. Take your pikk: Tumour viruses and dna damage response pathways. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160269. [Google Scholar] [CrossRef]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus dna replication and the host dna damage response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Warburton, A.; Markowitz, T.E.; Katz, J.P.; Pipas, J.M.; McBride, A.A. Recurrent integration of human papillomavirus genomes at transcriptional regulatory hubs. NPJ Genom. Med. 2021, 6, 101. [Google Scholar] [CrossRef]
- Feitelson, M.A.; Lee, J. Hepatitis b virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. 2007, 252, 157–170. [Google Scholar] [CrossRef]
- McBride, A.A. Human papillomaviruses: Diversity, infection and host interactions. Nat. Rev. Microbiol. 2022, 20, 95–108. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Shen, K.; McBride, A.A. Papillomavirus genomes associate with brd4 to replicate at fragile sites in the host genome. PLoS Pathog. 2014, 10, e1004117. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Maman, Y.; Jung, S.; Wong, N.; Callen, E.; Day, A.; Kieffer-Kwon, K.R.; Pekowska, A.; Zhang, H.; Rao, S.S.P.; et al. Genome organization drives chromosome fragility. Cell 2017, 170, 507–521.e518. [Google Scholar] [CrossRef] [PubMed]
- Barlow, J.H.; Faryabi, R.B.; Callén, E.; Wong, N.; Malhowski, A.; Chen, H.T.; Gutierrez-Cruz, G.; Sun, H.W.; McKinnon, P.; Wright, G.; et al. Identification of early replicating fragile sites that contribute to genome instability. Cell 2013, 152, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Canela, A.; Maman, Y.; Huang, S.N.; Wutz, G.; Tang, W.; Zagnoli-Vieira, G.; Callen, E.; Wong, N.; Day, A.; Peters, J.M.; et al. Topoisomerase ii-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol. Cell 2019, 75, 252–266.e258. [Google Scholar] [CrossRef]
- Majumder, K.; Wang, J.; Boftsi, M.; Fuller, M.S.; Rede, J.E.; Joshi, T.; Pintel, D.J. Parvovirus minute virus of mice interacts with sites of cellular dna damage to establish and amplify its lytic infection. Elife 2018, 7, e37750. [Google Scholar] [CrossRef]
- Majumder, K.; Boftsi, M.; Whittle, F.B.; Wang, J.; Fuller, M.S.; Joshi, T.; Pintel, D.J. The ns1 protein of the parvovirus mvm aids in the localization of the viral genome to cellular sites of dna damage. PLoS Pathog. 2020, 16, e1009002. [Google Scholar] [CrossRef]
- Schwartz, R.A.; Carson, C.T.; Schuberth, C.; Weitzman, M.D. Adeno-associated virus replication induces a dna damage response coordinated by dna-dependent protein kinase. J. Virol. 2009, 83, 6269–6278. [Google Scholar] [CrossRef]
- Adeyemi, R.O.; Pintel, D.J. The atr signaling pathway is disabled during infection with the parvovirus minute virus of mice. J. Virol. 2014, 88, 10189–10199. [Google Scholar] [CrossRef]
- Majumder, K.; Etingov, I.; Pintel, D.J. Protoparvovirus interactions with the cellular dna damage response. Viruses 2017, 9, 323. [Google Scholar] [CrossRef]
- Cater, J.E.; Pintel, D.J. The small non-structural protein ns2 of the autonomous parvovirus minute virus of mice is required for virus growth in murine cells. J. Gen. Virol. 1992, 73 Pt 7, 1839–1843. [Google Scholar] [CrossRef] [PubMed]
- Schoborg, R.V.; Pintel, D.J. Accumulation of mvm gene products is differentially regulated by transcription initiation, rna processing and protein stability. Virology 1991, 181, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Majumder, K.; Boftsi, M.; Pintel, D.J. Viral chromosome conformation capture (v3c) assays for identifying trans-interaction sites between lytic viruses and the cellular genome. Bio-Protoc. 2019, 9, e3198. [Google Scholar] [CrossRef] [PubMed]
- Boftsi, M.; Majumder, K.; Burger, L.R.; Pintel, D.J. Binding of ccctc-binding factor (ctcf) to the minute virus of mice genome is important for proper processing of viral p4-generated pre-mrnas. Viruses 2020, 12, 1368. [Google Scholar] [CrossRef] [PubMed]
- Bunke, L.E.; Larsen, C.I.S.; Pita-Aquino, J.N.; Jones, I.K.; Majumder, K. The dna damage sensor mre11 regulates efficient replication of the autonomous parvovirus minute virus of mice. J. Virol. 2023, e00461-23, online ahead of print. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of parp1 in dna repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Nüesch, J.P.; Rommelaere, J. Ns1 interaction with ckii alpha: Novel protein complex mediating parvovirus-induced cytotoxicity. J. Virol. 2006, 80, 4729–4739. [Google Scholar] [CrossRef]
- Liu, J.; Luo, S.; Zhao, H.; Liao, J.; Li, J.; Yang, C.; Xu, B.; Stern, D.F.; Xu, X.; Ye, K. Structural mechanism of the phosphorylation-dependent dimerization of the mdc1 forkhead-associated domain. Nucleic Acids Res. 2012, 40, 3898–3912. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Nüesch, J.P.; Tattersall, P. Asymmetric resolution of a parvovirus palindrome in vitro. J. Virol. 1993, 67, 1579–1589. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Nuesch, J.P.; Tattersall, P. In vitro excision and replication of 5’ telomeres of minute virus of mice dna from cloned palindromic concatemer junctions. Virology 1992, 190, 365–377. [Google Scholar] [CrossRef]
- Nüesch, J.P.; Rommelaere, J. A viral adaptor protein modulating casein kinase ii activity induces cytopathic effects in permissive cells. Proc. Natl. Acad. Sci. USA 2007, 104, 12482–12487. [Google Scholar] [CrossRef] [PubMed]
- Spycher, C.; Miller, E.S.; Townsend, K.; Pavic, L.; Morrice, N.A.; Janscak, P.; Stewart, G.S.; Stucki, M. Constitutive phosphorylation of mdc1 physically links the mre11-rad50-nbs1 complex to damaged chromatin. J. Cell Biol. 2008, 181, 227–240. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. Atm, atr, and dna-pk: The trinity at the heart of the dna damage response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Boftsi, M.; Whittle, F.B.; Wang, J.; Shepherd, P.; Burger, L.R.; Kaifer, K.A.; Lorson, C.L.; Joshi, T.; Pintel, D.J.; Majumder, K. The adeno-associated virus 2 (aav2) genome and rep 68/78 proteins interact with cellular sites of dna damage. Hum. Mol. Genet. 2021, 31, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Shah, G.A.; O’Shea, C.C. Viral and cellular genomes activate distinct dna damage responses. Cell 2015, 162, 987–1002. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target | Supplier | Catalog Number | Dosage |
|---|---|---|---|---|
| Olaparib | PARP | Selleckchem | S1060 | 1 µM |
| TDRL-505 | RPA binding | Millipore Sigma | 5.30535 | 50 µM |
| Caffeine | ATM and ATR | Millipore Sigma | W222402 | 2.5 µM |
| KU-55933 | ATM | Selleckchem | S1092 | 7 µM |
| Berzosertib | ATR | Selleckchem | S7102 | 1 µM and 2 µM |
| NU7441 (KU-57788) | DNA-PK | Selleckchem | S2638 | 10 µM |
| CAS 17374-26-4 | CK2 | Millipore Sigma | 218697 | 10 µM |
| CHIR-124 | CHK1 | Selleckchem | S2683 | 10 µM |
| Antibody | Supplier | Catalog Number | Application |
|---|---|---|---|
| Tubulin (Clone DM1A) | Sigma | 05-829 | WB |
| Mre11 | Cell Signaling | 4895 | WB |
| RPA phospho-Ser4, Ser8 | Thermo Fisher | A300-245A | IF |
| EXO1 phospho-Ser746 | Sigma | ABE1066 | IF |
| CHK1 phospho-Ser345 | Cell Signaling | 23415 | IF |
| CK2 phospho-Ser/Thr | Cell Signaling | 8738 | IF |
| DNAPKcs phospho-Ser2056 | Cell Signaling | 68716 | IF |
| Poly/Mono-ADP Ribose (E6F6A) | Cell Signaling | 83732 | IF |
| ATM phospho-Ser1981 | Cell Signaling | 13050 | IF |
| ATR phospho-Ser428 | Cell Signaling | 2853 | IF |
| NBS1 phospho-Ser95 | Cell Signaling | 3002 | IF |
| MDC1 phospho-T4 | Abcam | Ab35967 | IF |
| anti-Mouse-AF568 | Thermo Scientific | A11004 | IF |
| anti-Rabbit-AF488 | Thermo Scientific | A11034 | IF |
| Anti-mouse IgG, HRP-linked | Cell Signaling | 7076 | WB |
| Anti-rabbit IgG, HRP-linked | Cell Signaling | 7074 | WB |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Larsen, C.I.S.; Majumder, K. The Autonomous Parvovirus Minute Virus of Mice Localizes to Cellular Sites of DNA Damage Using ATR Signaling. Viruses 2023, 15, 1243. https://doi.org/10.3390/v15061243
Larsen CIS, Majumder K. The Autonomous Parvovirus Minute Virus of Mice Localizes to Cellular Sites of DNA Damage Using ATR Signaling. Viruses. 2023; 15(6):1243. https://doi.org/10.3390/v15061243
Chicago/Turabian StyleLarsen, Clairine I. S., and Kinjal Majumder. 2023. "The Autonomous Parvovirus Minute Virus of Mice Localizes to Cellular Sites of DNA Damage Using ATR Signaling" Viruses 15, no. 6: 1243. https://doi.org/10.3390/v15061243