
Engineering Adenoviral Vectors with Improved GBM Selectivity
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines and Adenoviral Vectors
2.2. Flow Cytometry
2.3. Viral Infection and Luciferase Assays
2.4. mRNA Expression Analysis
2.5. Statistical Analysis
3. Results
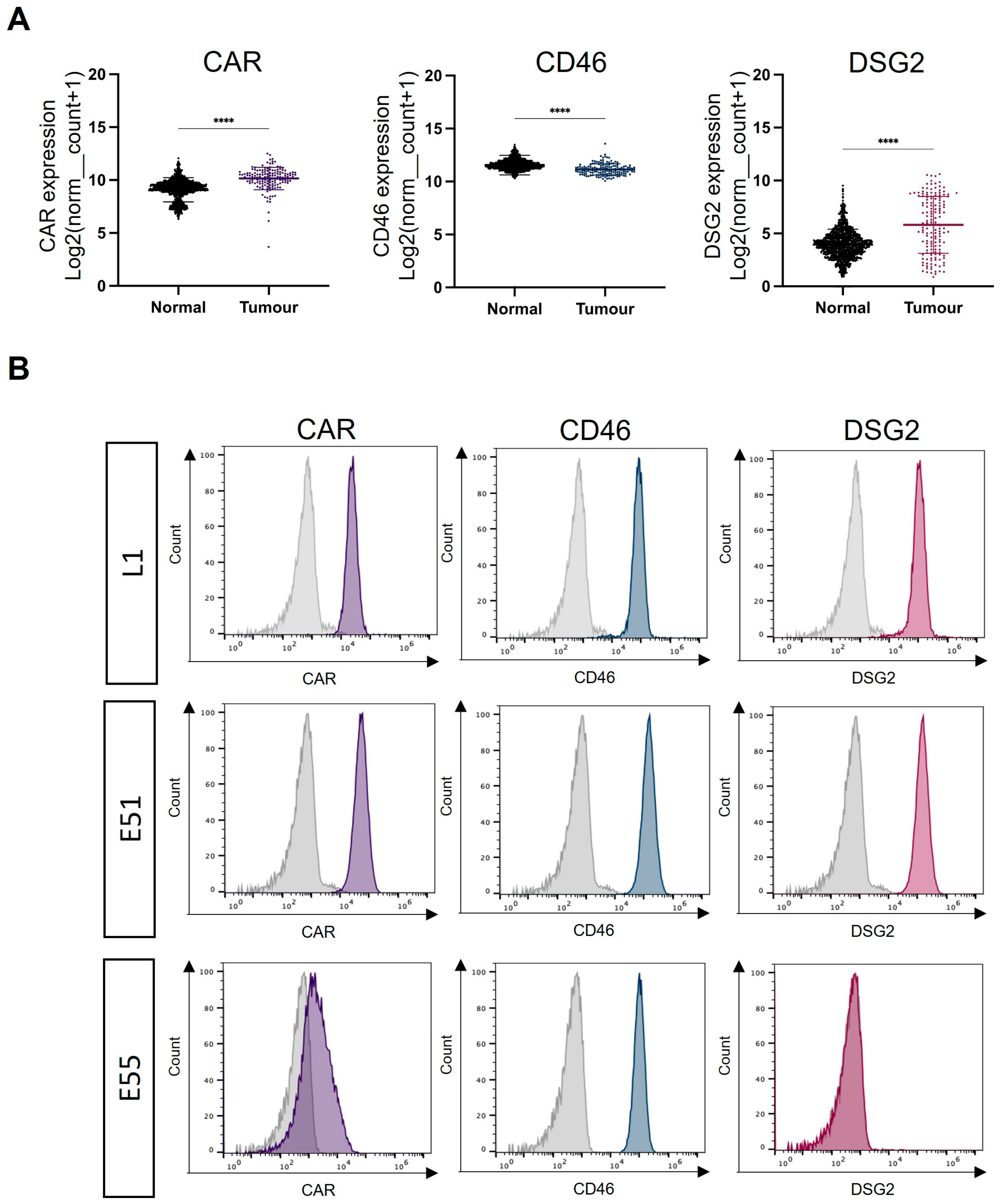
3.1. Expression of the Adenoviral Receptors CAR, CD46 and DSG2 in GBM
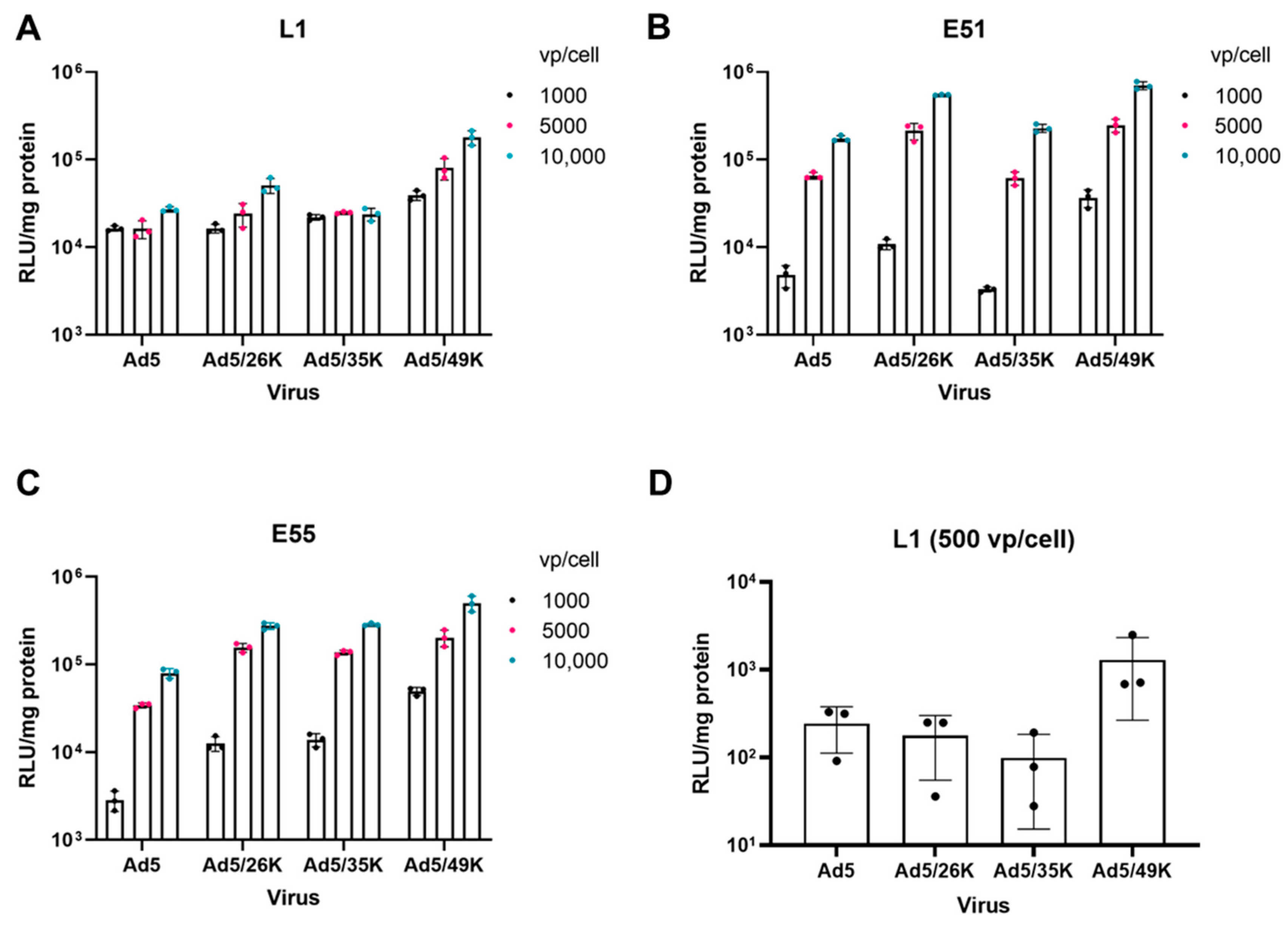
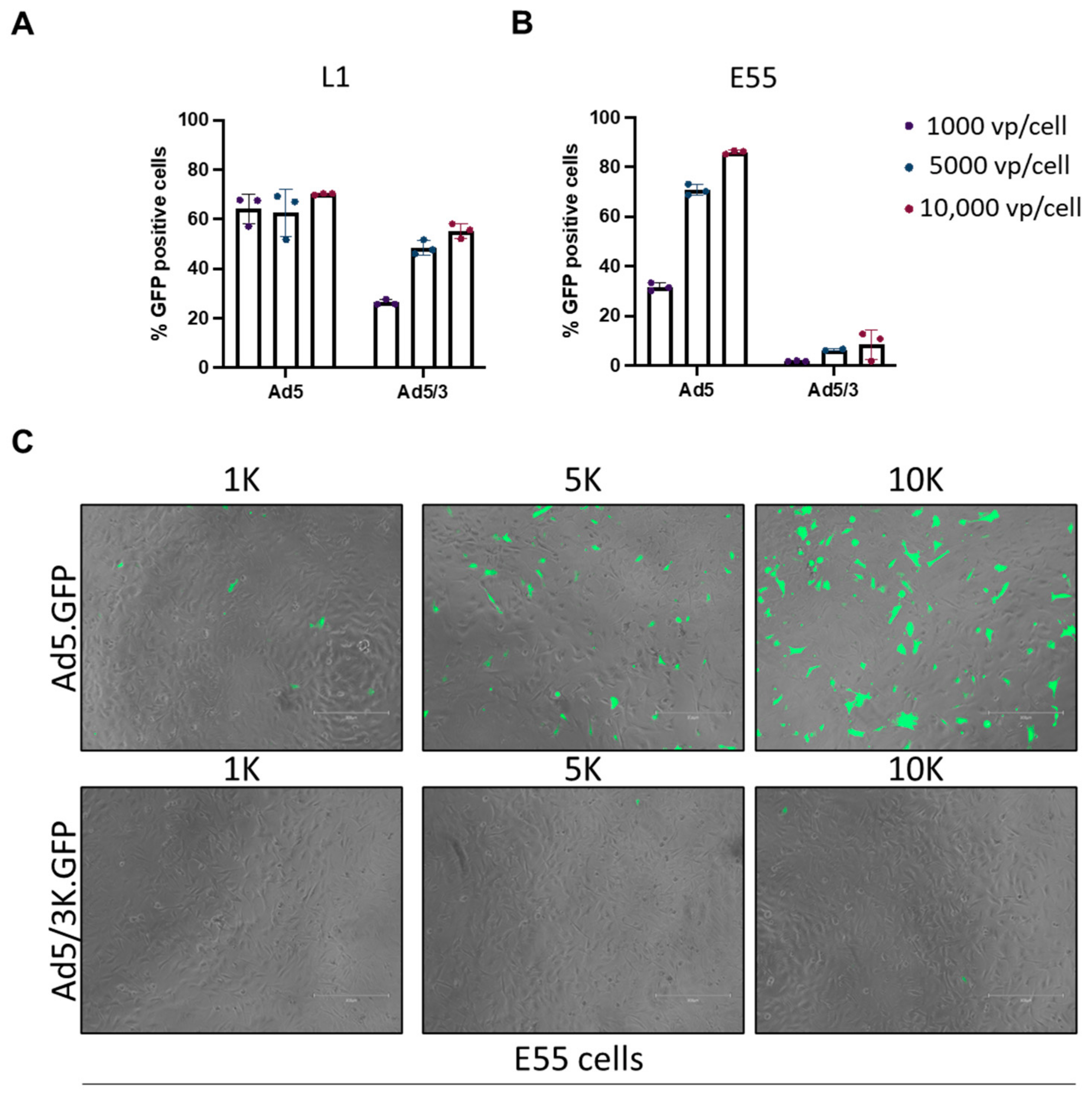
3.2. Transduction of Pseudotyped Adenoviral Vectors in GBM
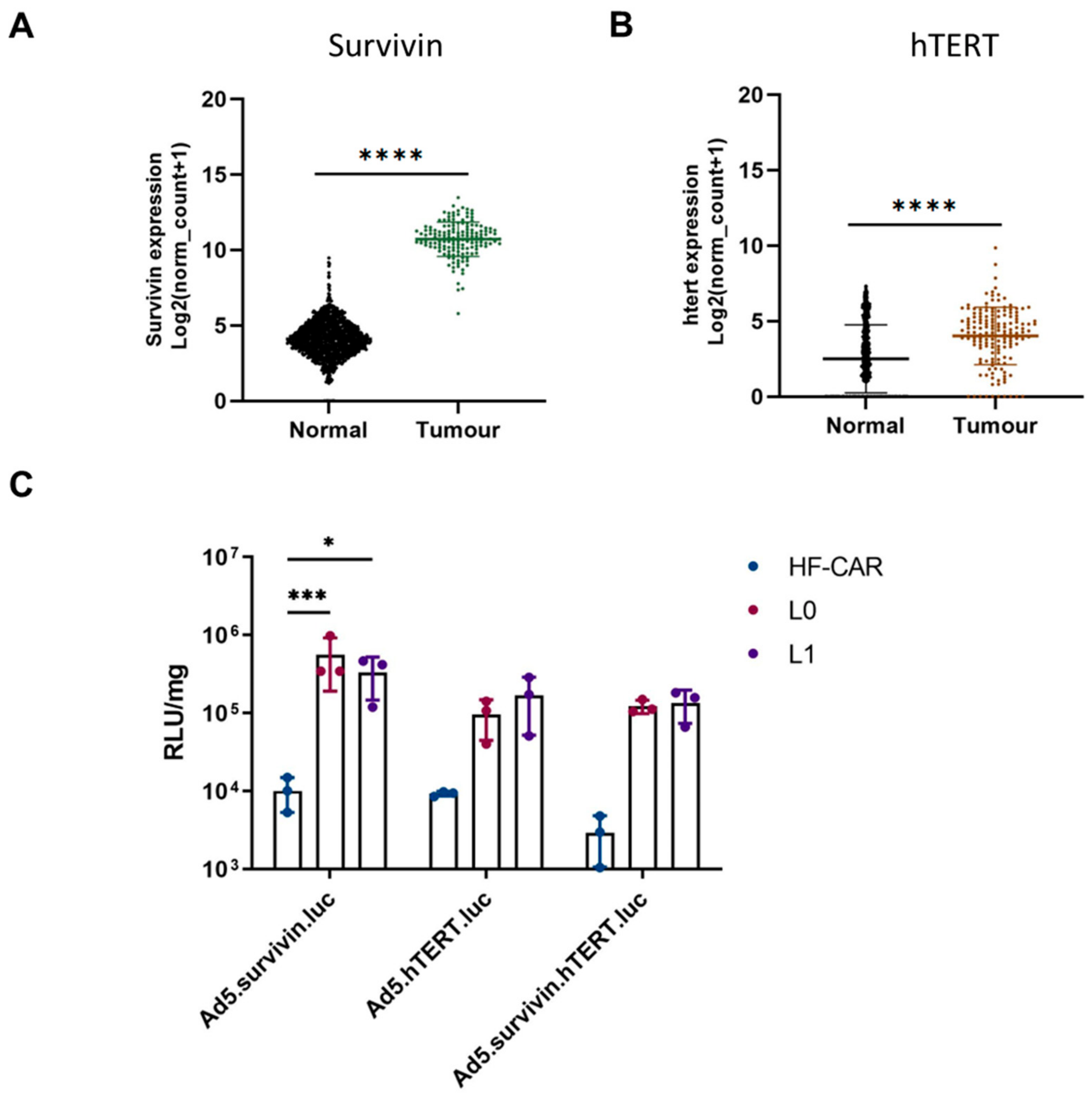
3.3. Tumour-Specific Promoter hTERT and Survivin Offer Benefits in GBM Cells over Normal Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 2014, 16 (Suppl. 4), iv1–iv63. [Google Scholar] [CrossRef] [PubMed]
- Di Carlo, D.T.; Cagnazzo, F.; Benedetto, N.; Morganti, R.; Perrini, P. Multiple high-grade gliomas: Epidemiology, management, and outcome: A systematic review and meta-analysis. Neurosurg. Rev. 2019, 42, 263–275. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Macedo, N.; Miller, D.M.; Haq, R.; Kaufman, H.L. Clinical landscape of oncolytic virus research in 2020. J. Immunother. Cancer 2020, 8, e001486. [Google Scholar] [CrossRef] [PubMed]
- Bett, A.J.; Prevec, L.; Graham, F.L. Packaging capacity and stability of human adenovirus type 5 vectors. J. Virol. 1993, 67, 5911–5921. [Google Scholar] [CrossRef]
- Sterman, D.H.; Treat, J.; Litzky, L.A.; Amin, K.M.; Coonrod, L.; Molnar-Kimber, K.; Recio, A.; Knox, L.; Wilson, J.M.; Albelda, S.M.; et al. Adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy in patients with localized malignancy: Results of a phase I clinical trial in malignant mesothelioma. Hum. Gene Ther. 1998, 9, 1083–1092. [Google Scholar] [CrossRef]
- Ram, Z.; Culver, K.W.; Oshiro, E.M.; Viola, J.J.; DeVroom, H.L.; Otto, E.; Long, Z.; Chiang, Y.; McGarrity, G.J.; Muul, L.M.; et al. Therapy of malignant brain tumors by intratumoral implantation of retroviral vector-producing cells. Nat. Med. 1997, 3, 1354–1361. [Google Scholar] [CrossRef]
- HAdV Working Group. Available online: http://hadvwg.gmu.edu/ (accessed on 29 March 2023).
- Parker, A.L.; Waddington, S.N.; Buckley, S.M.K.; Custers, J.; Havenga, M.J.E.; van Rooijen, N.; Goudsmit, J.; McVey, J.H.; Nicklin, S.A.; Baker, A.H. Effect of neutralizing sera on factor x-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009, 83, 479–483. [Google Scholar] [CrossRef]
- Mast, T.C.; Kierstead, L.; Gupta, S.B.; Nikas, A.A.; Kallas, E.G.; Novitsky, V.; Mbewe, B.; Pitisuttithum, P.; Schechter, M.; Vardas, E.; et al. International epidemiology of human pre-existing adenovirus (Ad) type-5, type-6, type-26 and type-36 neutralizing antibodies: Correlates of high Ad5 titers and implications for potential HIV vaccine trials. Vaccine 2010, 28, 950–957. [Google Scholar] [CrossRef]
- Bates, E.A.; Davies, J.A.; Váňová, J.; Nestić, D.; Meniel, V.S.; Koushyar, S.; Cunliffe, T.G.; Mundy, R.M.; Moses, E.; Uusi-Kerttula, H.K.; et al. Development of a low-seroprevalence, αvβ6 integrin-selective virotherapy based on human adenovirus type 10. Mol. Ther. Oncolytics 2022, 25, 43–56. [Google Scholar] [CrossRef]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junction. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef]
- Anders, M.; Vieth, M.; Röcken, C.; Ebert, M.; Pross, M.; Gretschel, S.; Schlag, P.M.; Wiedenmann, B.; Kemmner, W.; Höcker, M. Loss of the coxsackie and adenovirus receptor contributes to gastric cancer progression. Br. J. Cancer 2009, 100, 352–359. [Google Scholar] [CrossRef]
- Matsumoto, K.; Shariat, S.F.; Ayala, G.E.; Rauen, K.A.; Lerner, S.P. Loss of coxsackie and adenovirus receptor expression is associated with features of aggressive bladder cancer. Urology 2005, 66, 441–446. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Gaggar, A.; Ni, S.; Li, Z.-Y.; Lieber, A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005, 79, 7478–7491. [Google Scholar] [CrossRef]
- Waddington, S.N.; McVey, J.H.; Bhella, D.; Parker, A.L.; Barker, K.; Atoda, H.; Pink, R.; Buckley, S.M.K.; Greig, J.A.; Denby, L.; et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell 2008, 132, 397–409. [Google Scholar] [CrossRef]
- Parker, A.L.; Waddington, S.N.; Nicol, C.G.; Shayakhmetov, D.M.; Buckley, S.M.; Denby, L.; Kemball-Cook, G.; Ni, S.; Lieber, A.; McVey, J.H.; et al. Multiple vitamin K-dependent coagulation zymogens promote adenovirus-mediated gene delivery to hepatocytes. Blood 2006, 108, 2554–2561. [Google Scholar] [CrossRef]
- Carlisle, R.C.; Di, Y.; Cerny, A.M.; Sonnen, A.F.-P.; Sim, R.B.; Green, N.K.; Subr, V.; Ulbrich, K.; Gilbert, R.J.C.; Fisher, K.D.; et al. Human erythrocytes bind and inactivate type 5 adenovirus by presenting Coxsackie virus-adenovirus receptor and complement receptor 1. Blood 2009, 113, 1909–1918. [Google Scholar] [CrossRef]
- Lang, F.F.; Conrad, C.; Gomez-Manzano, C.; Yung, W.K.A.; Sawaya, R.; Weinberg, J.S.; Prabhu, S.S.; Rao, G.; Fuller, G.N.; Aldape, K.D.; et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2018, 36, 1419–1427. [Google Scholar] [CrossRef]
- Baker, A.T.; Mundy, R.M.; Davies, J.A.; Rizkallah, P.J.; Parker, A.L. Human adenovirus type 26 uses sialic acid-bearing glycans as a primary cell entry receptor. Sci. Adv. 2019, 5, eaax3567. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, Z.-Y.; Liu, Y.; Persson, J.; Beyer, I.; Möller, T.; Koyuncu, D.; Drescher, M.R.; Strauss, R.; Zhang, X.-B.; et al. Desmoglein 2 is a receptor for adenovirus serotypes 3, 7, 11 and 14. Nat. Med. 2011, 17, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Rawal, P.; Zhao, L. Sialometabolism in Brain Health and Alzheimer’s Disease. Front. Neurosci. 2021, 15, 648617. Available online: https://www.frontiersin.org/articles/10.3389/fnins.2021.648617 (accessed on 27 March 2023). [CrossRef] [PubMed]
- Van Aarsen, L.A.K.; Leone, D.R.; Ho, S.; Dolinski, B.M.; McCoon, P.E.; LePage, D.J.; Kelly, R.; Heaney, G.; Rayhorn, P.; Reid, C.; et al. Antibody-mediated blockade of integrin alpha v beta 6 inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res. 2008, 68, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Uusi-Kerttula, H.; Davies, J.A.; Thompson, J.M.; Wongthida, P.; Evgin, L.; Shim, K.G.; Bradshaw, A.; Baker, A.T.; Rizkallah, P.J.; Jones, R.; et al. Ad5NULL-A20: A Tropism-Modified, αvβ6 Integrin-Selective Oncolytic Adenovirus for Epithelial Ovarian Cancer Therapies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 4215–4224. [Google Scholar] [CrossRef]
- Davies, J.A.; Marlow, G.; Uusi-Kerttula, H.K.; Seaton, G.; Piggott, L.; Badder, L.M.; Clarkson, R.W.E.; Chester, J.D.; Parker, A.L. Efficient Intravenous Tumor Targeting Using the αvβ6 Integrin-Selective Precision Virotherapy Ad5NULL-A20. Viruses 2021, 13, 864. [Google Scholar] [CrossRef]
- Uusi-Kerttula, H.; Davies, J.; Coughlan, L.; Hulin-Curtis, S.; Jones, R.; Hanna, L.; Chester, J.D.; Parker, A.L. Pseudotyped αvβ6 integrin-targeted adenovirus vectors for ovarian cancer therapies. Oncotarget 2016, 7, 27926–27937. [Google Scholar] [CrossRef]
- Piccirillo, S.G.M.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761–765. [Google Scholar] [CrossRef]
- Hoang-Minh, L.B.; Siebzehnrubl, F.A.; Yang, C.; Suzuki-Hatano, S.; Dajac, K.; Loche, T.; Andrews, N.; Schmoll Massari, M.; Patel, J.; Amin, K.; et al. Infiltrative and drug-resistant slow-cycling cells support metabolic heterogeneity in glioblastoma. EMBO J. 2018, 37, e98772. [Google Scholar] [CrossRef]
- Siebzehnrubl, F.A.; Silver, D.J.; Tugertimur, B.; Deleyrolle, L.P.; Siebzehnrubl, D.; Sarkisian, M.R.; Devers, K.G.; Yachnis, A.T.; Kupper, M.D.; Neal, D.; et al. The ZEB1 pathway links glioblastoma initiation, invasion and chemoresistance. EMBO Mol. Med. 2013, 5, 1196–1212. [Google Scholar] [CrossRef]
- Pollard, S.M.; Yoshikawa, K.; Clarke, I.D.; Danovi, D.; Stricker, S.; Russell, R.; Bayani, J.; Head, R.; Lee, M.; Bernstein, M.; et al. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 2009, 4, 568–580. [Google Scholar] [CrossRef]
- The AdZ Adenovirus Cloning System|AdZ.cf.ac.uk. Available online: https://adz.cf.ac.uk/ (accessed on 27 March 2023).
- Uusi-Kerttula, H.; Legut, M.; Davies, J.; Jones, R.; Hudson, E.; Hanna, L.; Stanton, R.J.; Chester, J.D.; Parker, A.L. Incorporation of Peptides Targeting EGFR and FGFR1 into the Adenoviral Fiber Knob Domain and Their Evaluation as Targeted Cancer Therapies. Hum. Gene Ther. 2015, 26, 320–329. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repečka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Baker, A.T.; Davies, J.A.; Bates, E.A.; Moses, E.; Mundy, R.M.; Marlow, G.; Cole, D.K.; Bliss, C.M.; Rizkallah, P.J.; Parker, A.L. The Fiber Knob Protein of Human Adenovirus Type 49 Mediates Highly Efficient and Promiscuous Infection of Cancer Cell Lines Using a Novel Cell Entry Mechanism. J. Virol. 2021, 95, e01849-20. [Google Scholar] [CrossRef]
- Dakin, R.S.; Parker, A.L.; Delles, C.; Nicklin, S.A.; Baker, A.H. Efficient transduction of primary vascular cells by the rare adenovirus serotype 49 vector. Hum. Gene Ther. 2015, 26, 312–319. [Google Scholar] [CrossRef]
- Nguyen, H.N.; Lie, A.; Li, T.; Chowdhury, R.; Liu, F.; Ozer, B.; Wei, B.; Green, R.M.; Ellingson, B.M.; Wang, H.-J.; et al. Human TERT promoter mutation enables survival advantage from MGMT promoter methylation in IDH1 wild-type primary glioblastoma treated by standard chemoradiotherapy. Neuro-Oncology 2017, 19, 394–404. [Google Scholar] [CrossRef]
- Das, A.; Tan, W.-L.; Teo, J.; Smith, D.R. Expression of survivin in primary glioblastomas. J. Cancer Res. Clin. Oncol. 2002, 128, 302–306. [Google Scholar] [CrossRef]
- Alekseenko, I.V.; Pleshkan, V.V.; Kopantzev, E.P.; Stukacheva, E.A.; Chernov, I.P.; Vinogradova, T.V.; Sverdlov, E.D. Activity of the Upstream Component of Tandem TERT/Survivin Promoters Depends on Features of the Downstream Component. PLoS ONE 2012, 7, e46474. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Bates, E.A.; Counsell, J.R.; Alizert, S.; Baker, A.T.; Suff, N.; Boyle, A.; Bradshaw, A.C.; Waddington, S.N.; Nicklin, S.A.; Baker, A.H.; et al. In Vitro and In Vivo Evaluation of Human Adenovirus Type 49 as a Vector for Therapeutic Applications. Viruses 2021, 13, 1483. [Google Scholar] [CrossRef]
- Stepanenko, A.A.; Sosnovtseva, A.O.; Valikhov, M.P.; Chernysheva, A.A.; Cherepanov, S.A.; Yusubalieva, G.M.; Ruzsics, Z.; Lipatova, A.V.; Chekhonin, V.P. Superior infectivity of the fiber chimeric oncolytic adenoviruses Ad5/35 and Ad5/3 over Ad5-delta-24-RGD in primary glioma cultures. Mol. Ther. Oncolytics 2022, 24, 230–248. [Google Scholar] [CrossRef] [PubMed]
- Ter Horst, M.; Brouwer, E.; Verwijnen, S.; Rodijk, M.; de Jong, M.; Hoeben, R.; de Leeuw, B.; Smitt, P.S. Targeting malignant gliomas with a glial fibrillary acidic protein (GFAP)-selective oncolytic adenovirus. J. Gene Med. 2007, 9, 1071–1079. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bates, E.A.; Lovatt, C.; Plein, A.R.; Davies, J.A.; Siebzehnrubl, F.A.; Parker, A.L. Engineering Adenoviral Vectors with Improved GBM Selectivity. Viruses 2023, 15, 1086. https://doi.org/10.3390/v15051086
Bates EA, Lovatt C, Plein AR, Davies JA, Siebzehnrubl FA, Parker AL. Engineering Adenoviral Vectors with Improved GBM Selectivity. Viruses. 2023; 15(5):1086. https://doi.org/10.3390/v15051086
Chicago/Turabian StyleBates, Emily A., Charlotte Lovatt, Alice R. Plein, James A. Davies, Florian A. Siebzehnrubl, and Alan L. Parker. 2023. "Engineering Adenoviral Vectors with Improved GBM Selectivity" Viruses 15, no. 5: 1086. https://doi.org/10.3390/v15051086