
Targeting the Host Mitochondria as a Novel Human Cytomegalovirus Antiviral Strategy

Abstract
:1. Introduction
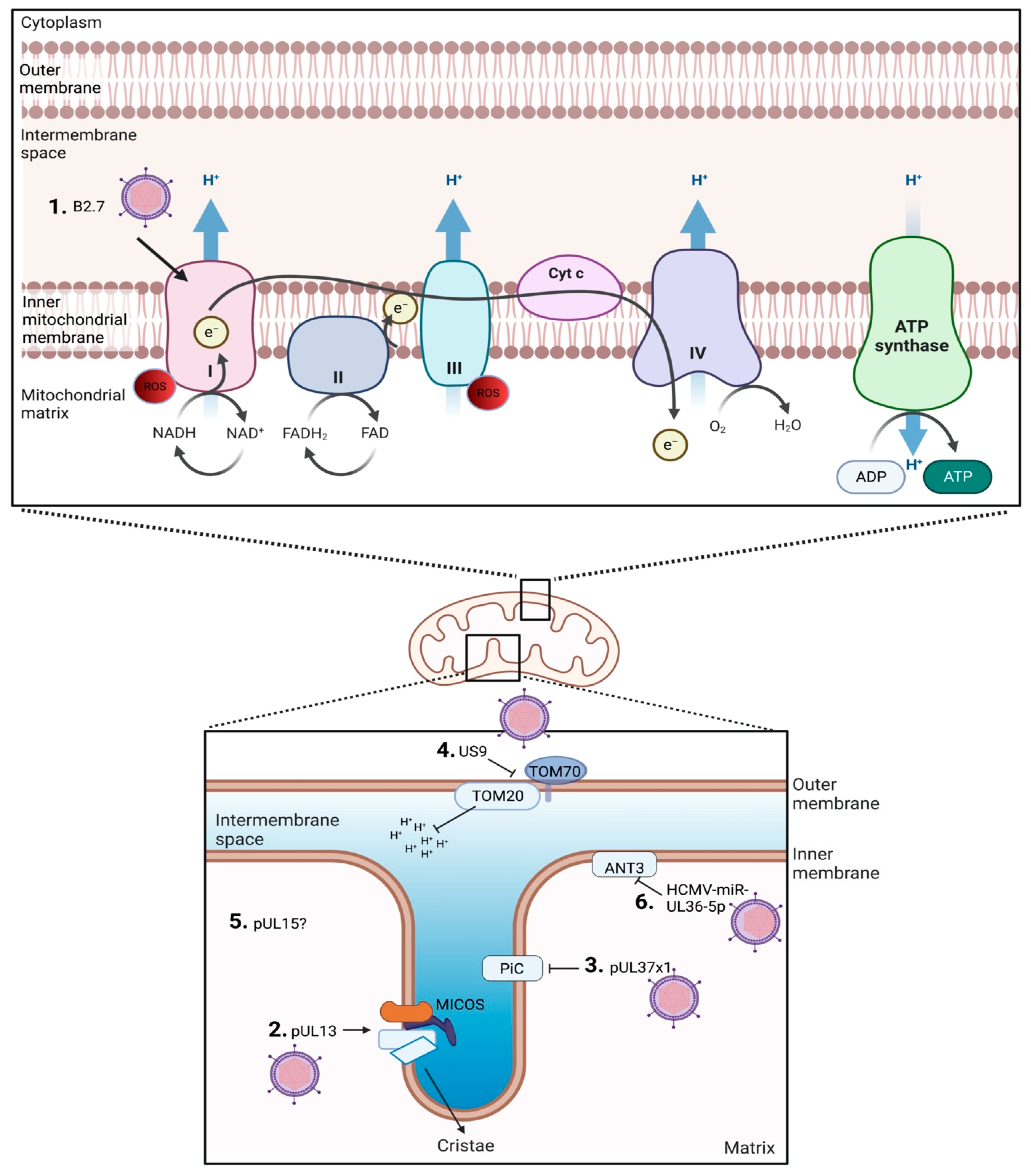
2. HCMV Targets Host Mitochondria
3. ETC and Its Role in HCMV Replication
3.1. ETC Complex I: NADH-Ubiquinone Oxidoreductase
3.2. ETC Complex II: Succinate Dehydrogenase
3.3. ETC Complex III: CoQ-Cytochrome c Reductase
3.4. ETC Complex IV: Cytochrome c Oxidase
3.5. Complex V: F1F0 ATPase Synthase
3.6. ETC-Associated Mechanisms
4. Mitochondrial Morphology and Its Role in CMV Replication
5. Prospective HCMV Antivirals and Respective Mode of Action

{kind=link}
| Drug | Target | Results | Notes | Reference |
|---|---|---|---|---|
| MitoTam | ETC complex I | - Increased ROS - Decreased mitochondrial membrane potential | No change in glycolysis or ATP levels | [77] |
| ME-143 ME-344 | ETC complex I | - Decreased mitochondrial membrane potential | ME-344 induced ETC complex disruption | [78] |
| γ-tocotrienol | ETC complexes I and II | - Increased ROS | [79] | |
| MitoVES | ETC complex II (proposed) | - Increased ROS - Decreased mitochondrial membrane potential | Decreased effect on non-malignant cells | [80] |
| CycT | OHPHOS | - Increased mitochondrial membrane potential - Increased ROS - Increased mitochondrial fission | [83] | |
| - Decreased OXPHOS and expression of ETC subunits | Independent of hedgehog signaling | [97] | ||
| Metformin | ETC complex I | - Increased glycolysis - Uncoupled respiration | [91] | |
| - Decreased ETC function | Requires increased MMP | [90] | ||
| Not described | - Decreased TCA intermediates | [89] | ||
| - Decreased TCA intermediates and carnitines | Increased aspartate release | [88] | ||
| mDIVI1 | DRP1 | - Increased ROS | [92] |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thaker, S.K.; Ch’ng, J.; Christofk, H.R. Viral hijacking of cellular metabolism. BMC Biol. 2019, 17, 59. [Google Scholar] [CrossRef] [PubMed]
- Pant, A.; Dsouza, L.; Yang, Z. Alteration in Cellular Signaling and Metabolic Reprogramming during Viral Infection. mBio 2021, 12, e0063521. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.M.; Xu, S.; Munger, J. Stealing the Keys to the Kitchen: Viral Manipulation of the Host Cell Metabolic Network. Trends Microbiol. 2015, 23, 789–798. [Google Scholar] [CrossRef] [PubMed]
- Shenk, T.; Alwine, J.C. Human Cytomegalovirus: Coordinating Cellular Stress, Signaling, and Metabolic Pathways. Annu. Rev. Virol. 2014, 1, 355–374. [Google Scholar] [CrossRef]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 2011, 85, 1573–1580. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. ChREBP, a glucose-responsive transcriptional factor, enhances glucose metabolism to support biosynthesis in human cytomegalovirus-infected cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1951–1956. [Google Scholar] [CrossRef]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human Cytomegalovirus Alters Host Cell Mitochondrial Function during Acute Infection. J. Virol. 2020, 94, e01183-19. [Google Scholar] [CrossRef]
- Rodriguez-Sanchez, I.; Schafer, X.L.; Monaghan, M.; Munger, J. The Human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog. 2019, 15, e1007569. [Google Scholar] [CrossRef]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef]
- McArdle, J.; Schafer, X.L.; Munger, J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J. Virol. 2011, 85, 705–714. [Google Scholar] [CrossRef]
- Chambers, J.W.; Maguire, T.G.; Alwine, J.C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 2010, 84, 1867–1873. [Google Scholar] [CrossRef]
- Betsinger, C.N.; Jankowski, C.S.R.; Hofstadter, W.A.; Federspiel, J.D.; Otter, C.J.; Jean Beltran, P.M.; Cristea, I.M. The human cytomegalovirus protein pUL13 targets mitochondrial cristae architecture to increase cellular respiration during infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101675118. [Google Scholar] [CrossRef]
- Kaarbo, M.; Ager-Wick, E.; Osenbroch, P.O.; Kilander, A.; Skinnes, R.; Muller, F.; Eide, L. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 2011, 11, 935–945. [Google Scholar] [CrossRef]
- Karniely, S.; Weekes, M.P.; Antrobus, R.; Rorbach, J.; van Haute, L.; Umrania, Y.; Smith, D.L.; Stanton, R.J.; Minczuk, M.; Lehner, P.J.; et al. Human Cytomegalovirus Infection Upregulates the Mitochondrial Transcription and Translation Machineries. mBio 2016, 7, e00029. [Google Scholar] [CrossRef]
- Federspiel, J.D.; Cook, K.C.; Kennedy, M.A.; Venkatesh, S.S.; Otter, C.J.; Hofstadter, W.A.; Jean Beltran, P.M.; Cristea, I.M. Mitochondria and Peroxisome Remodeling across Cytomegalovirus Infection Time Viewed through the Lens of Inter-ViSTA. Cell Rep. 2020, 32, 107943. [Google Scholar] [CrossRef]
- Koyuncu, E.; Purdy, J.G.; Rabinowitz, J.D.; Shenk, T. Saturated very long chain fatty acids are required for the production of infectious human cytomegalovirus progeny. PLoS Pathog. 2013, 9, e1003333. [Google Scholar] [CrossRef]
- Purdy, J.G.; Shenk, T.; Rabinowitz, J.D. Fatty acid elongase 7 catalyzes lipidome remodeling essential for human cytomegalovirus replication. Cell Rep. 2015, 10, 1375–1385. [Google Scholar] [CrossRef]
- Spencer, C.M.; Schafer, X.L.; Moorman, N.J.; Munger, J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011, 85, 5814–5824. [Google Scholar] [CrossRef]
- Combs, J.A.; Monk, C.H.; Harrison, M.A.A.; Norton, E.B.; Morris, C.A.; Sullivan, D.E.; Zwezdaryk, K.J. Inhibiting cytomegalovirus replication through targeting the host electron transport chain. Antiviral Res. 2021, 194, 105159. [Google Scholar] [CrossRef]
- Monk, C.H.; Zwezdaryk, K.J. Host Mitochondrial Requirements of Cytomegalovirus Replication. Curr. Clin. Microbiol. Rep. 2020, 7, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tyl, M.D.; Betsinger, C.N.; Cristea, I.M. Virus-host protein interactions as footprints of human cytomegalovirus replication. Curr. Opin. Virol. 2022, 52, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Youle, R.J. The role of mitochondria in apoptosis. Annu. Rev. Genet. 2009, 43, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Fletcher-Etherington, A.; Nobre, L.; Nightingale, K.; Antrobus, R.; Nichols, J.; Davison, A.J.; Stanton, R.J.; Weekes, M.P. Human cytomegalovirus protein pUL36: A dual cell death pathway inhibitor. Proc. Natl. Acad. Sci. USA 2020, 117, 18771–18779. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Kim, J.H.; Nogalski, M.T.; Stevenson, E.V.; Chan, G.C.; Caskey, J.R.; Cieply, S.J.; Yurochko, A.D. Human Cytomegalovirus Promotes Survival of Infected Monocytes via a Distinct Temporal Regulation of Cellular Bcl-2 Family Proteins. J. Virol. 2015, 90, 2356–2371. [Google Scholar] [CrossRef]
- Norris, K.L.; Youle, R.J. Cytomegalovirus proteins vMIA and m38.5 link mitochondrial morphogenesis to Bcl-2 family proteins. J. Virol. 2008, 82, 6232–6243. [Google Scholar] [CrossRef]
- Brune, W.; Andoniou, C.E. Die Another Day: Inhibition of Cell Death Pathways by Cytomegalovirus. Viruses 2017, 9, 249. [Google Scholar] [CrossRef]
- Ren, Z.; Zhang, X.; Ding, T.; Zhong, Z.; Hu, H.; Xu, Z.; Deng, J. Mitochondrial Dynamics Imbalance: A Strategy for Promoting Viral Infection. Front. Microbiol. 2020, 11, 1992. [Google Scholar] [CrossRef]
- McCormick, A.L.; Smith, V.L.; Chow, D.; Mocarski, E.S. Disruption of mitochondrial networks by the human cytomegalovirus UL37 gene product viral mitochondrion-localized inhibitor of apoptosis. J. Virol. 2003, 77, 631–641. [Google Scholar] [CrossRef]
- Jean Beltran, P.M.; Mathias, R.A.; Cristea, I.M. A Portrait of the Human Organelle Proteome In Space and Time during Cytomegalovirus Infection. Cell Syst. 2016, 3, 361–373.e6. [Google Scholar] [CrossRef]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I binding by a virally encoded RNA regulates mitochondria-induced cell death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef]
- Perera, M.R.; Roche, K.L.; Murphy, E.A.; Sinclair, J.H. A Viral Long Non-Coding RNA Protects against Cell Death during Human Cytomegalovirus Infection of CD14+ Monocytes. Viruses 2022, 14, 246. [Google Scholar] [CrossRef]
- Poncet, D.; Pauleau, A.L.; Szabadkai, G.; Vozza, A.; Scholz, S.R.; Le Bras, M.; Briere, J.J.; Jalil, A.; Le Moigne, R.; Brenner, C.; et al. Cytopathic effects of the cytomegalovirus-encoded apoptosis inhibitory protein vMIA. J. Cell Biol. 2006, 174, 985–996. [Google Scholar] [CrossRef]
- Goldmacher, V.S.; Bartle, L.M.; Skaletskaya, A.; Dionne, C.A.; Kedersha, N.L.; Vater, C.A.; Han, J.W.; Lutz, R.J.; Watanabe, S.; Cahir McFarland, E.D.; et al. A cytomegalovirus-encoded mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl. Acad. Sci. USA 1999, 96, 12536–12541. [Google Scholar] [CrossRef]
- Choi, H.J.; Park, A.; Kang, S.; Lee, E.; Lee, T.A.; Ra, E.A.; Lee, J.; Lee, S.; Park, B. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun. 2018, 9, 125. [Google Scholar] [CrossRef]
- Guo, X.; Huang, Y.; Qi, Y.; Liu, Z.; Ma, Y.; Shao, Y.; Jiang, S.; Sun, Z.; Ruan, Q. Human cytomegalovirus miR-UL36-5p inhibits apoptosis via downregulation of adenine nucleotide translocator 3 in cultured cells. Arch. Virol. 2015, 160, 2483–2490. [Google Scholar] [CrossRef]
- Wang, N.; Ma, Y.; Li, M.; Gao, S.; Wang, L.; Qi, Y.; Ji, Y.; Sun, Z.; Ruan, Q. Transcription characteristics of the human cytomegalovirus UL13 gene. Arch. Virol. 2013, 158, 473–477. [Google Scholar] [CrossRef]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Hertel, L.; Mocarski, E.S. Global analysis of host cell gene expression late during cytomegalovirus infection reveals extensive dysregulation of cell cycle gene expression and induction of Pseudomitosis independent of US28 function. J. Virol. 2004, 78, 11988–12011. [Google Scholar] [CrossRef]
- Rodenburg, R.J. Mitochondrial complex I-linked disease. Biochim. Biophys. Acta 2016, 1857, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Hock, D.H.; Robinson, D.R.L.; Stroud, D.A. Blackout in the powerhouse: Clinical phenotypes associated with defects in the assembly of OXPHOS complexes and the mitoribosome. Biochem. J. 2020, 477, 4085–4132. [Google Scholar] [CrossRef] [PubMed]
- Kameli, R.; Amanat, M.; Rezaei, Z.; Hosseionpour, S.; Nikbakht, S.; Alizadeh, H.; Ashrafi, M.R.; Omrani, A.; Garshasbi, M.; Tavasoli, A.R. RNASET2-deficient leukoencephalopathy mimicking congenital CMV infection and Aicardi-Goutieres syndrome: A case report with a novel pathogenic variant. Orphanet J. Rare Dis. 2019, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Kim, Y.S. Malonate metabolism: Biochemistry, molecular biology, physiology, and industrial application. J. Biochem. Mol. Biol. 2002, 35, 443–451. [Google Scholar] [CrossRef]
- Akopova, O.; Kolchinskaya, L.; Nosar, V.; Mankovska, I.; Sagach, V. Diazoxide affects mitochondrial bioenergetics by the opening of mKATP channel on submicromolar scale. BMC Mol. Cell Biol. 2020, 21, 31. [Google Scholar] [CrossRef]
- Wojtovich, A.P.; Brookes, P.S. The endogenous mitochondrial complex II inhibitor malonate regulates mitochondrial ATP-sensitive potassium channels: Implications for ischemic preconditioning. Biochim. Biophys. Acta 2008, 1777, 882–889. [Google Scholar] [CrossRef]
- Speir, E.; Shibutani, T.; Yu, Z.X.; Ferrans, V.; Epstein, S.E. Role of reactive oxygen intermediates in cytomegalovirus gene expression and in the response of human smooth muscle cells to viral infection. Circ. Res. 1996, 79, 1143–1152. [Google Scholar] [CrossRef]
- Frizzell, N.; William, L. Bioenergetics and Oxidative Metabolism. In Medical Biochemistry; Elsevier: Amsterdam, The Netherlands, 2019; Volume 5, pp. 93–110. [Google Scholar]
- Vempati, U.D.; Diaz, F.; Barrientos, A.; Narisawa, S.; Mian, A.M.; Millán, J.L.; Boise, L.H.; Moraes, C.T. Role of cytochrome C in apoptosis: Increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome C release. Mol. Cell. Biol. 2007, 27, 1771–1783. [Google Scholar] [CrossRef]
- Claus, C.; Schonefeld, K.; Hubner, D.; Chey, S.; Reibetanz, U.; Liebert, U.G. Activity increase in respiratory chain complexes by rubella virus with marginal induction of oxidative stress. J. Virol. 2013, 87, 8481–8492. [Google Scholar] [CrossRef]
- Tripathy, M.K.; Mitra, D. Differential modulation of mitochondrial OXPHOS system during HIV-1 induced T-cell apoptosis: Up regulation of Complex-IV subunit COX-II and its possible implications. Apoptosis 2010, 15, 28–40. [Google Scholar] [CrossRef]
- Maruf, A.; Lee, O.; O’Brien, P. Modifications of Mitochondrial Function by Toxicants. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar] [CrossRef]
- Goldmacher, V.S. vMIA, a viral inhibitor of apoptosis targeting mitochondria. Biochimie 2002, 84, 177–185. [Google Scholar] [CrossRef]
- Landini, M.P.; Rugolo, M. Increased accumulation of a lipophilic cation (tetraphenylphosphonium) in human embryo fibroblasts after infection with cytomegalovirus. J. Gen. Virol. 1984, 65 Pt 12, 2269–2272. [Google Scholar] [CrossRef]
- Hong, C.T.; Chau, K.Y.; Schapira, A.H. The Cytomegalovirus protein pUL37x1 targets mitochondria to mediate neuroprotection. Sci. Rep. 2016, 6, 31373. [Google Scholar] [CrossRef]
- Crowe, W.E.; Maglova, L.M.; Ponka, P.; Russell, J.M. Human cytomegalovirus-induced host cell enlargement is iron dependent. Am. J. Physiol. Cell Physiol. 2004, 287, C1023–C1030. [Google Scholar] [CrossRef]
- Wild, M.; Hahn, F.; Grau, B.; Herrmann, L.; Niesar, A.; Schutz, M.; Lorion, M.M.; Ackermann, L.; Tsogoeva, S.B.; Marschall, M. The Artemisinin-Derived Autofluorescent Compound BG95 Exerts Strong Anticytomegaloviral Activity Based on a Mitochondrial Targeting Mechanism. Int. J. Mol. Sci. 2020, 21, 5578. [Google Scholar] [CrossRef]
- Hutterer, C.; Niemann, I.; Milbradt, J.; Frohlich, T.; Reiter, C.; Kadioglu, O.; Bahsi, H.; Zeittrager, I.; Wagner, S.; Einsiedel, J.; et al. The broad-spectrum antiinfective drug artesunate interferes with the canonical nuclear factor kappa B (NF-kappaB) pathway by targeting RelA/p65. Antiviral Res. 2015, 124, 101–109. [Google Scholar] [CrossRef]
- Hahn, F.; Frohlich, T.; Frank, T.; Bertzbach, L.D.; Kohrt, S.; Kaufer, B.B.; Stamminger, T.; Tsogoeva, S.B.; Marschall, M. Artesunate-derived monomeric, dimeric and trimeric experimental drugs—Their unique mechanistic basis and pronounced antiherpesviral activity. Antiviral Res. 2018, 152, 104–110. [Google Scholar] [CrossRef]
- Wild, M.; Bertzbach, L.D.; Tannig, P.; Wangen, C.; Muller, R.; Herrmann, L.; Frohlich, T.; Tsogoeva, S.B.; Kaufer, B.B.; Marschall, M.; et al. The trimeric artesunate derivative TF27 exerts strong anti-cytomegaloviral efficacy: Focus on prophylactic efficacy and oral treatment of immunocompetent mice. Antiviral Res. 2020, 178, 104788. [Google Scholar] [CrossRef]
- Luo, C.L.; Chen, X.P.; Li, L.L.; Li, Q.Q.; Li, B.X.; Xue, A.M.; Xu, H.F.; Dai, D.K.; Shen, Y.W.; Tao, L.Y.; et al. Poloxamer 188 attenuates in vitro traumatic brain injury-induced mitochondrial and lysosomal membrane permeabilization damage in cultured primary neurons. J. Neurotrauma 2013, 30, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Ramzan, R.; Wen, L.; Vogt, S. New extension of the Mitchell Theory for oxidative phosphorylation in mitochondria of living organisms. Biochim. Biophys. Acta 2010, 1800, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Ramzan, R.; Moosdorf, R.; Vogt, S. The role of mitochondrial membrane potential in ischemic heart failure. Mitochondrion 2011, 11, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Harrison, M.A.A.; Hochreiner, E.M.; Benjamin, B.P.; Lawler, S.E.; Zwezdaryk, K.J. Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses 2022, 14, 103. [Google Scholar] [CrossRef]
- Sharon-Friling, R.; Goodhouse, J.; Colberg-Poley, A.M.; Shenk, T. Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc. Natl. Acad. Sci. USA 2006, 103, 19117–19122. [Google Scholar] [CrossRef]
- Poncet, D.; Larochette, N.; Pauleau, A.L.; Boya, P.; Jalil, A.A.; Cartron, P.F.; Vallette, F.; Schnebelen, C.; Bartle, L.M.; Skaletskaya, A.; et al. An anti-apoptotic viral protein that recruits Bax to mitochondria. J. Biol. Chem. 2004, 279, 22605–22614. [Google Scholar] [CrossRef]
- Karbowski, M.; Norris, K.L.; Cleland, M.M.; Jeong, S.Y.; Youle, R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature 2006, 443, 658–662. [Google Scholar] [CrossRef]
- Arnoult, D.; Bartle, L.M.; Skaletskaya, A.; Poncet, D.; Zamzami, N.; Park, P.U.; Sharpe, J.; Youle, R.J.; Goldmacher, V.S. Cytomegalovirus cell death suppressor vMIA blocks Bax- but not Bak-mediated apoptosis by binding and sequestering Bax at mitochondria. Proc. Natl. Acad. Sci. USA 2004, 101, 7988–7993. [Google Scholar] [CrossRef]
- Murray, L.A.; Sheng, X.; Cristea, I.M. Orchestration of protein acetylation as a toggle for cellular defense and virus replication. Nat. Commun. 2018, 9, 4967. [Google Scholar] [CrossRef]
- Sheng, X.; Cristea, I.M. The antiviral sirtuin 3 bridges protein acetylation to mitochondrial integrity and metabolism during human cytomegalovirus infection. PLoS Pathog. 2021, 17, e1009506. [Google Scholar] [CrossRef]
- Cui, Q.; Wen, S.; Huang, P. Targeting cancer cell mitochondria as a therapeutic approach: Recent updates. Future Med. Chem. 2017, 9, 929–949. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Cheng, G.; Hardy, M.; Ouari, O.; Lopez, M.; Joseph, J.; Zielonka, J.; Dwinell, M.B. A review of the basics of mitochondrial bioenergetics, metabolism, and related signaling pathways in cancer cells: Therapeutic targeting of tumor mitochondria with lipophilic cationic compounds. Redox Biol. 2018, 14, 316–327. [Google Scholar] [CrossRef]
- Rohlenova, K.; Sachaphibulkij, K.; Stursa, J.; Bezawork-Geleta, A.; Blecha, J.; Endaya, B.; Werner, L.; Cerny, J.; Zobalova, R.; Goodwin, J.; et al. Selective Disruption of Respiratory Supercomplexes as a New Strategy to Suppress Her2(high) Breast Cancer. Antioxid. Redox Signal. 2017, 26, 84–103. [Google Scholar] [CrossRef]
- Lim, S.C.; Carey, K.T.; McKenzie, M. Anti-cancer analogues ME-143 and ME-344 exert toxicity by directly inhibiting mitochondrial NADH: Ubiquinone oxidoreductase (Complex I). Am. J. Cancer Res. 2015, 5, 689–701. [Google Scholar]
- Wang, H.; Luo, J.; Tian, W.; Yan, W.; Ge, S.; Zhang, Y.; Sun, W. gamma-Tocotrienol inhibits oxidative phosphorylation and triggers apoptosis by inhibiting mitochondrial complex I subunit NDUFB8 and complex II subunit SDHB. Toxicology 2019, 417, 42–53. [Google Scholar] [CrossRef]
- Dong, L.F.; Jameson, V.J.; Tilly, D.; Prochazka, L.; Rohlena, J.; Valis, K.; Truksa, J.; Zobalova, R.; Mahdavian, E.; Kluckova, K.; et al. Mitochondrial targeting of alpha-tocopheryl succinate enhances its pro-apoptotic efficacy: A new paradigm for effective cancer therapy. Free Radic. Biol. Med. 2011, 50, 1546–1555. [Google Scholar] [CrossRef]
- Dong, L.F.; Low, P.; Dyason, J.C.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef]
- Cheng, G.; Zielonka, J.; Dranka, B.P.; McAllister, D.; Mackinnon, A.C., Jr.; Joseph, J.; Kalyanaraman, B. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012, 72, 2634–2644. [Google Scholar] [CrossRef]
- Alam, M.M.; Sohoni, S.; Kalainayakan, S.P.; Garrossian, M.; Zhang, L. Cyclopamine tartrate, an inhibitor of Hedgehog signaling, strongly interferes with mitochondrial function and suppresses aerobic respiration in lung cancer cells. BMC Cancer 2016, 16, 150. [Google Scholar] [CrossRef]
- Sohoni, S.; Ghosh, P.; Wang, T.; Kalainayakan, S.P.; Vidal, C.; Dey, S.; Konduri, P.C.; Zhang, L. Elevated Heme Synthesis and Uptake Underpin Intensified Oxidative Metabolism and Tumorigenic Functions in Non-Small Cell Lung Cancer Cells. Cancer Res. 2019, 79, 2511–2525. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.; Pollak, M.N.; Hirst, J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Ben Sahra, I.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Romero, I.L.; Litchfield, L.M.; Lengyel, E.; Locasale, J.W. Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab. 2016, 24, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Peiris-Pages, M.; Bonuccelli, G.; Sotgia, F.; Lisanti, M.P. Mitochondrial fission as a driver of stemness in tumor cells: mDIVI1 inhibits mitochondrial function, cell migration and cancer stem cell (CSC) signalling. Oncotarget 2018, 9, 13254–13275. [Google Scholar] [CrossRef]
- Dong, L.; Neuzil, J. Targeting mitochondria as an anticancer strategy. Cancer Commun. 2019, 39, 63. [Google Scholar] [CrossRef]
- Vasan, K.; Werner, M.; Chandel, N.S. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020, 32, 341–352. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Roth, K.G.; Mambetsariev, I.; Kulkarni, P.; Salgia, R. The Mitochondrion as an Emerging Therapeutic Target in Cancer. Trends Mol. Med. 2020, 26, 119–134. [Google Scholar] [CrossRef]
- Kalainayakan, S.P.; Ghosh, P.; Dey, S.; Fitzgerald, K.E.; Sohoni, S.; Konduri, P.C.; Garrossian, M.; Liu, L.; Zhang, L. Cyclopamine tartrate, a modulator of hedgehog signaling and mitochondrial respiration, effectively arrests lung tumor growth and progression. Sci. Rep. 2019, 9, 1405. [Google Scholar] [CrossRef]
- Barnes, L.L.; Capuano, A.W.; Aiello, A.E.; Turner, A.D.; Yolken, R.H.; Torrey, E.F.; Bennett, D.A. Cytomegalovirus infection and risk of Alzheimer disease in older black and white individuals. J. Infect. Dis. 2015, 211, 230–237. [Google Scholar] [CrossRef]
- Stragliotto, G.; Rahbar, A.; Solberg, N.W.; Lilja, A.; Taher, C.; Orrego, A.; Bjurman, B.; Tammik, C.; Skarman, P.; Peredo, I.; et al. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: A randomized, double-blind, hypothesis-generating study. Int. J. Cancer 2013, 133, 1204–1213. [Google Scholar] [CrossRef]
- Jia, Y.J.; Liu, J.; Han, F.F.; Wan, Z.R.; Gong, L.L.; Liu, H.; Zhang, W.; Wardell, T.; Lv, Y.L.; Liu, L.H. Cytomegalovirus infection and atherosclerosis risk: A meta-analysis. J. Med. Virol. 2017, 89, 2196–2206. [Google Scholar] [CrossRef]
- Samson, L.D.; van den Berg, S.P.; Engelfriet, P.; Boots, A.M.; Hendriks, M.; de Rond, L.G.; de Zeeuw-Brouwer, M.L.; Verschuren, W.M.; Borghans, J.A.; Buisman, A.M.; et al. Limited effect of duration of CMV infection on adaptive immunity and frailty: Insights from a 27-year-long longitudinal study. Clin. Transl. Immunol. 2020, 9, e1193. [Google Scholar] [CrossRef]
- Foster, H.; Piper, K.; DePledge, L.; Li, H.F.; Scanlan, J.; Jae-Guen, Y.; Boeckh, M.; Cobbs, C. Human cytomegalovirus seropositivity is associated with decreased survival in glioblastoma patients. Neurooncol. Adv. 2019, 1, vdz020. [Google Scholar] [CrossRef]

| Drug | Mitochondrial Target | Result | Reference |
|---|---|---|---|
| Rotenone | ETC Complex I | 1 log reduction in HCMV titer (TR stain) No change in HCMV titer (Towne strain) Significant cell death reported | [20] |
| Decreased HCMV titer, increased oxidative stress, decreased ATP No change in cell death reported | [32] | ||
| Metformin | ETC Complex I | 1 log reduction in HCMV titer 2.5 log reduction in HCMV titer (reduced glucose conditions) No change in cell death reported | [20] |
| Antimycin A | ETC Complex III | 2 log reduction in HCMV titer (TR strain) 0.5 log reduction in HCMV titer (Towne strain) No change in cell death reported | [20] |
| Oligomycin | ATP Synthase | 1 log reduction in HCMV titer (TR strain) No change in HCMV titer (Towne strain) No change in cell death reported | [20] |
| [14] | |||
| FCCP | ETC | No significant change in HCMV titer No change in cell death reported | [20] |
| Chloramphenicol | ETC | Reduced HCMV titers Increased cell death reported | [14] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bachman, L.O.; Zwezdaryk, K.J. Targeting the Host Mitochondria as a Novel Human Cytomegalovirus Antiviral Strategy. Viruses 2023, 15, 1083. https://doi.org/10.3390/v15051083
Bachman LO, Zwezdaryk KJ. Targeting the Host Mitochondria as a Novel Human Cytomegalovirus Antiviral Strategy. Viruses. 2023; 15(5):1083. https://doi.org/10.3390/v15051083
Chicago/Turabian StyleBachman, Lauryn O., and Kevin J. Zwezdaryk. 2023. "Targeting the Host Mitochondria as a Novel Human Cytomegalovirus Antiviral Strategy" Viruses 15, no. 5: 1083. https://doi.org/10.3390/v15051083