
The Involvement of Ubiquitination and SUMOylation in Retroviruses Infection and Latency

Abstract
:1. Introduction
2. Ubiquitination and SUMOylation Pathways
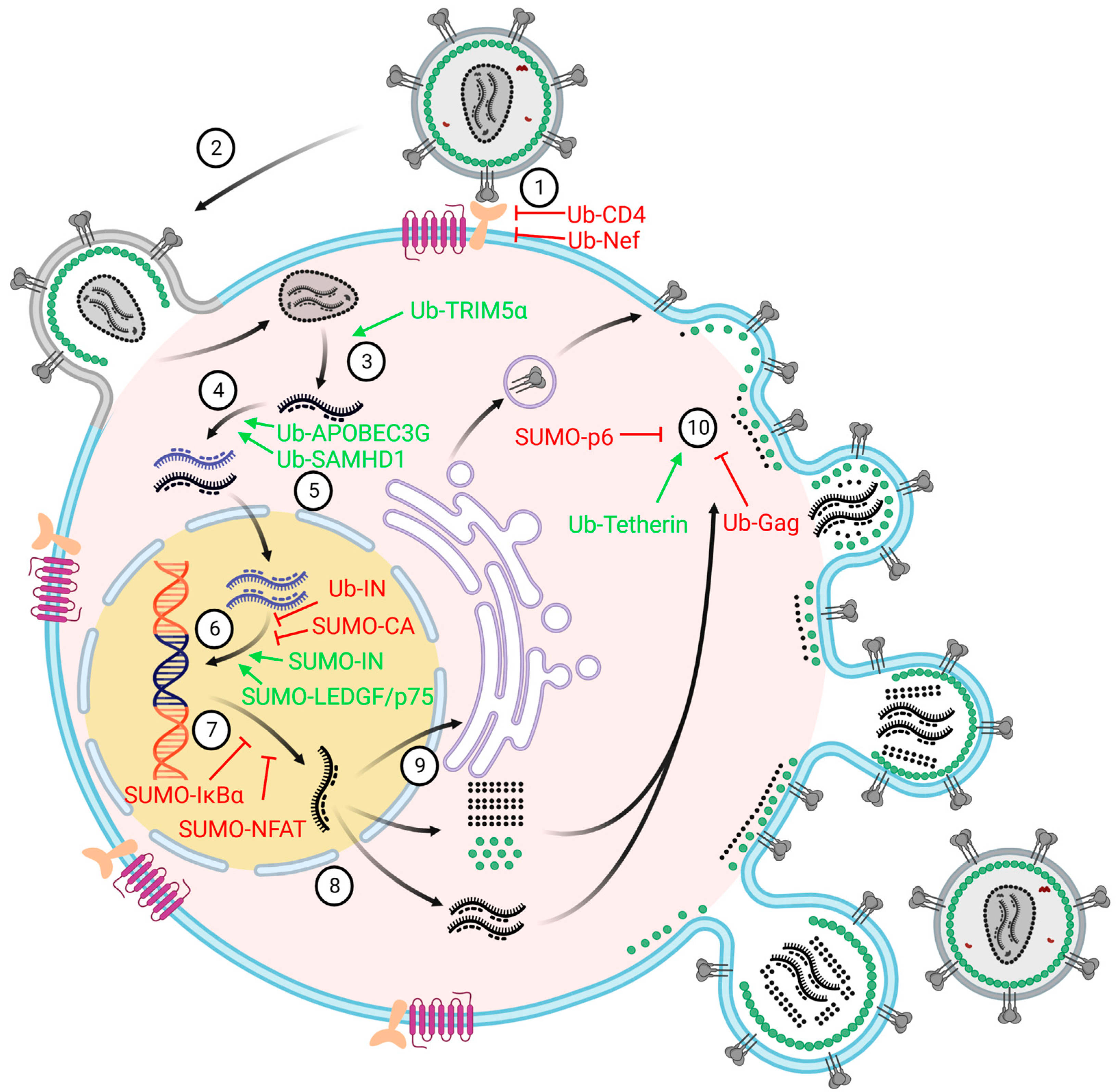
3. The Involvement of Ubiquitination and SUMOylation in Retroviruses Infection
3.1. Ubiquitination and Retroviruses Infection
3.2. SUMOylation and Retroviruses Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTMs | Virus (s) | Viral Target (s) | Cellular Target (s) | Function | References |
|---|---|---|---|---|---|
| Ubiquitination | HIV-1 | - | CD4 | Inhibit viral entry and avoid super-infection | [87] |
| HIV-1 | Nef | - | Inhibit viral entry and avoid super-infection | [89] | |
| HIV-1 | - | TRIM5α | Accelerate viral uncoating | [98] | |
| HIV-1 | - | TAK1 | Induce IFN production and pro-inflammatory cytokines secretion against HIV-1 infection | [102] | |
| HIV-1, MLV | - | APOBEC3G | Promote nascent viral single-stranded cDNA synthesis | [106] | |
| HIV-1 | - | SAMHD1 | Promote the synthesis of the viral genomic DNA | [117] | |
| HIV-1, MLV | IN | - | Suppress viral DNA integration and prevent provirus formation | [121] | |
| HIV-1, SIV, MLV, ALV | Gag | - | Interfere the virion release of multiple retroviruses | [130,136] | |
| HIV-1 | - | Tetherin | Promote viral release | [139,140] | |
| SUMOylation | HIV-1 | p6 | - | Decrease viral infectivity | [149] |
| HIV-1, MoMuLV | IN | - | Ensure efficient infectivity | [154] | |
| MoMuLV | CA | - | Required for early events of viral infection and the formation of proviruses | [158] | |
| HTLV-1 | Tax | - | Activate NF-κB pathway | [160,164] | |
| HIV-1 | - | LEDGF/p75 | Promote efficient viral integration | [169] | |
| HIV-1 | - | IκBα | Suppress NF-κB-activated viral genes | [172] | |
| HIV-1 | - | NFAT | Promote nuclear localization of NFAT and silence NFAT-targeted genes | [81,174] | |
| ERV | - | TRIM28 | Deposit SETDB1 and hnRNP K on ERV | [177] | |
| ERV | - | Morc3 | Bind Daxx and promote H3.3 deposition on ERV | [178] |
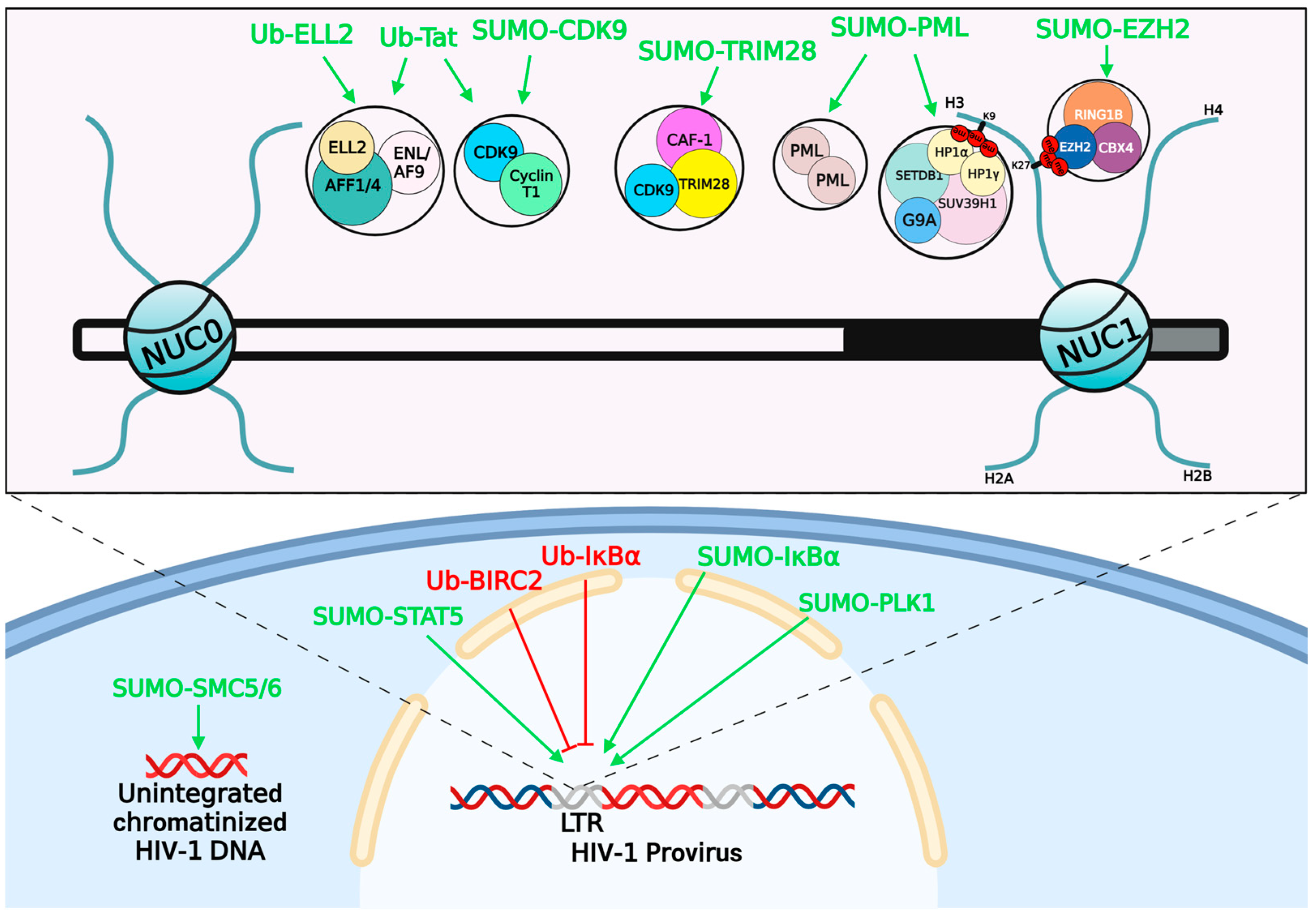
4. The Involvement of Ubiquitination and SUMOylation in Retroviruses Latency
4.1. Ubiquitination and Retroviruses Latency
4.2. SUMOylation and Retroviruses Latency
| PTMs | Virus (s) | Viral Target (s) | Cellular Target (s) | Function | References |
|---|---|---|---|---|---|
| Ubiquitination | HIV-1 | Tat | - | Disrupt HIV-1 transcription elongation | [187] |
| HIV-1 | - | ELL2 | Disrupt HIV-1 transcription | [192] | |
| HIV-1 | - | BIRC2 | Activate NF–κB signaling and reactivate HIV-1 transcription | [22] | |
| HIV-1 | - | IκBα | Reactivate HIV-1 transcription | [193] | |
| HTLV-1 | Tax | - | Activate NF–κB pathway by E2 UBC13-mediated K63-Ub | [195] | |
| HTLV-1 | Tax | - | Degrade Tax by E3 PDLIM2-mediated K48-Ub | [196] | |
| HTLV-1 | p13 | - | Interfere Tax-CBP/p300 interaction and inhibit proviral transcription | [197] | |
| HTLV-1 | - | H2A | Inhibit provirus reactivation from latency | [201] | |
| SUMOylation | HIV-1 | - | CDK9 | Reduce RNAP II activation to suppress HIV-1 transcription | [204] |
| HIV-1 | - | TRIM28 | Maintain HIV-1 latency by coalescing with CAF-1 | [205] | |
| HIV-1 | - | PLK1 | Prevent cell death of HIV-1-infected cells and increase the viral latent reservoir | [208] | |
| HIV-1 | - | STAT5 | Inhibit its nuclear translocation and promote HIV-1 latency | [211,212] | |
| HIV-1 | - | IκBα | Enhance the hijacking activity of IκBα to NF–κB and impair HIV-1 transcription | [214] | |
| HIV-1 | - | PML | Degrade PML via ubiquitin-proteasome pathway in HIV-1 productively infected cells | [216] | |
| HIV-1 | - | SMC5/6 | Silence integration-competent HIV-1 proviruses | [217] | |
| HIV-1 | - | EZH2 | Mediate the formation of H3K27me3 and suppress HIV-1 transcription | [218] |
5. Ubiquitination- and SUMOylation-Targeted Anti-Retroviral Drugs
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sefton, B.M.; Beemon, K.; Hunter, T. Comparison of the expression of the src gene of Rous sarcoma virus in vitro and in vivo. J. Virol. 1978, 28, 957–971. [Google Scholar] [CrossRef] [PubMed]
- Mikkers, H.; Berns, A. Retroviral insertional mutagenesis:Tagging cancer pathways. Adv. Cancer Res. 2003, 88, 53–99. [Google Scholar] [PubMed]
- Persistent, generalized lymphadenopathy among homosexual males. Morb. Mortal Wkly. Rep. 1982, 31, 249–251.
- Agarwal-Jans, S. Timeline: HIV. Cell 2020, 183, 550. [Google Scholar] [CrossRef]
- Khanal, S.; Schank, M.; El Gazzar, M.; Moorman, J.P.; Yao, Z.Q. HIV-1 Latency and Viral Reservoirs: Existing Reversal Approaches and Potential Technologies, Targets, and Pathways Involved in HIV Latency Studies. Cells 2021, 10, 475. [Google Scholar] [CrossRef]
- MacLachlan, N.J.; Dubovi, E.J. (Eds.) Chapter 14—Retroviridae. In Fenner’s Veterinary Virology, 5th ed.; Academic Press: Boston, MA, USA, 2017; pp. 269–297. [Google Scholar]
- Campbell, E.M.; Hope, T.J. Live cell imaging of the HIV-1 life cycle. Trends Microbiol. 2008, 16, 580–587. [Google Scholar] [CrossRef]
- Wilen, C.B.; Tilton, J.C.; Doms, R.W. HIV: Cell Binding and Entry. Cold Spring Harb. Perspect. Med. 2012, 2, a006866. [Google Scholar] [CrossRef]
- Arhel, N. Revisiting HIV-1 uncoating. Retrovirology 2010, 7, 96. [Google Scholar] [CrossRef]
- Hughes, S.H. Reverse Transcription of Retroviruses and LTR Retrotransposons. Microbiol. Spectr. 2015, 3, MDNA3-0027-2014. [Google Scholar] [CrossRef]
- Hindmarsh, P.; Leis, J. Retroviral DNA Integration. Microbiol. Mol. Biol. Rev. 1999, 63, 836–843. [Google Scholar] [CrossRef]
- Blissenbach, M.; Grewe, B.; Hoffmann, B.; Brandt, S.; Überla, K. Nuclear RNA Export and Packaging Functions of HIV-1 Rev Revisited. J. Virol. 2010, 84, 6598–6604. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, S.; Batisse, J.; Libre, C.; Bernacchi, S.; Marquet, R.; Paillart, J.-C. HIV-1 Replication and the Cellular Eukaryotic Translation Apparatus. Viruses 2015, 7, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef] [PubMed]
- Sarabia, I.; Bosque, A. HIV-1 Latency and Latency Reversal: Does Subtype Matter? Viruses 2019, 11, 1104. [Google Scholar] [CrossRef]
- van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B. Establishment and molecular mechanisms of HIV-1 latency in T cells. Curr. Opin. Virol. 2013, 3, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Judge, M.; Parker, E.; Naniche, D.; Souëf, P.L. Gene Expression: The Key to Understanding HIV-1 Infection? Microbiol. Mol. Biol. Rev. 2020, 84, e00080-19. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Battivelli, E.; Verdin, E. Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med. 2015, 66, 407–421. [Google Scholar] [CrossRef]
- Zhang, X.; Ma, X.; Jing, S.; Zhang, H.; Zhang, Y. Non-coding RNAs and retroviruses. Retrovirology 2018, 15, 20. [Google Scholar] [CrossRef]
- Balvay, L.; Lastra, M.L.; Sargueil, B.; Darlix, J.-L.; Ohlmann, T. Translational control of retroviruses. Nat. Rev. Microbiol. 2007, 5, 128–140. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Li, J.; Chen, C.; Ma, X.; Geng, G.; Liu, B.; Zhang, Y.; Zhang, S.; Zhong, F.; Liu, C.; Yin, Y.; et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016, 7, 11730. [Google Scholar] [CrossRef] [PubMed]
- Kincaid, R.P.; Burke, J.M.; Sullivan, C.S. RNA virus microRNA that mimics a B-cell oncomiR. Proc. Natl. Acad. Sci. USA 2012, 109, 3077–3082. [Google Scholar] [CrossRef] [PubMed]
- Saayman, S.; Ackley, A.; Turner, A.-M.W.; Famiglietti, M.; Bosque, A.; Clemson, M.; Planelles, V.; Morris, K.V. An HIV-Encoded Antisense Long Noncoding RNA Epigenetically Regulates Viral Transcription. Mol. Ther. 2014, 22, 1164–1175. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef]
- Ramazi, S.; Zahiri, J. Post-translational modifications in proteins: Resources, tools and prediction methods. Database 2021, 2021, baab012. [Google Scholar] [CrossRef]
- Kumar, R.; Mehta, D.; Mishra, N.; Nayak, D.; Sunil, S. Role of Host-Mediated Post-Translational Modifications (PTMs) in RNA Virus Pathogenesis. Int. J. Mol. Sci. 2021, 22, 323. [Google Scholar] [CrossRef]
- Leis, J.; Johnson, S.; Collins, L.S.; Traugh, J.A. Effects of phosphorylation of avian retrovirus nucleocapsid protein pp12 on binding of viral RNA. J. Biol. Chem. 1984, 259, 7726–7732. [Google Scholar] [CrossRef]
- Nguyen, K.; Das, B.; Dobrowolski, C.; Karn, J. Multiple Histone Lysine Methyltransferases Are Required for the Establishment and Maintenance of HIV-1 Latency. mBio 2017, 8, e00133-17. [Google Scholar] [CrossRef]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG Methylation Controls Reactivation of HIV from Latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef]
- Kaehlcke, K.; Dorr, A.; Hetzer-Egger, C.; Kiermer, V.; Henklein, P.; Schnoelzer, M.; Loret, E.; Cole, P.A.; Verdin, E.; Ott, M. Acetylation of Tat Defines a CyclinT1-Independent Step in HIV Transactivation. Mol. Cell 2003, 12, 167–176. [Google Scholar] [CrossRef]
- Ott, M.; Schnölzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.-R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Diedrich, J.K.; Kulp, D.W.; Pauthner, M.; He, L.; Park, S.-K.R.; Sok, D.; Su, C.Y.; Delahunty, C.M.; Menis, S.; et al. Global site-specific N-glycosylation analysis of HIV envelope glycoprotein. Nat. Commun. 2017, 8, 14954. [Google Scholar] [CrossRef] [PubMed]
- Rousso, I.; Mixon, M.B.; Chen, B.K.; Kim, P.S. Palmitoylation of the HIV-1 envelope glycoprotein is critical for viral infectivity. Proc. Natl. Acad. Sci. USA 2000, 97, 13523–13525. [Google Scholar] [CrossRef] [PubMed]
- González, S.A.; Paladino, M.G.; Affranchino, J.L. Palmitoylation of the feline immunodeficiency virus envelope glycoprotein and its effect on fusion activity and envelope incorporation into virions. Virology 2012, 428, 1–10. [Google Scholar] [CrossRef]
- Bryant, M.; Ratner, L. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA 1990, 87, 523–527. [Google Scholar] [CrossRef]
- Pal, R.; Reitz, M.S., Jr.; Tschachler, E.; Gallo, R.C.; Sarngadharan, M.G.; Veronese, F.D. Myristoylation of gag Proteins of HIV-1 Plays an Important Role in Virus Assembly. AIDS Res. Hum. Retrovir. 1990, 6, 721–730. [Google Scholar] [CrossRef]
- Shoji, S.; Tashiro, A.; Kubota, Y. Antimyristoylation of gag Proteins in Human T-Cell Leukemia and Human Immunodeficiency Viruses with N-Myristoyl Glycinal Diethylacetal1. J. Biochem. 1988, 103, 747–749. [Google Scholar] [CrossRef]
- Edwards, D.C.; McKinnon, K.M.; Fenizia, C.; Jung, K.-J.; Brady, J.N.; Pise-Masison, C.A. Inhibition of Geranylgeranyl Transferase-I Decreases Cell Viability of HTLV-1-Transformed Cells. Viruses 2011, 3, 1815–1835. [Google Scholar] [CrossRef]
- Farzan, M.; Mirzabekov, T.; Kolchinsky, P.; Wyatt, R.; Cayabyab, M.; Gerard, N.P.; Gerard, C.; Sodroski, J.; Choe, H. Tyrosine Sulfation of the Amino Terminus of CCR5 Facilitates HIV-1 Entry. Cell 1999, 96, 667–676. [Google Scholar] [CrossRef]
- Cimbro, R.; Gallant, T.R.; Dolan, M.A.; Guzzo, C.; Zhang, P.; Lin, Y.; Miao, H.; Van Ryk, D.; Arthos, J.; Gorshkova, I.; et al. Tyrosine sulfation in the second variable loop (V2) of HIV-1 gp120 stabilizes V2–V3 interaction and modulates neutralization sensitivity. Proc. Natl. Acad. Sci. USA 2014, 111, 3152–3157. [Google Scholar] [CrossRef] [PubMed]
- Ravid, T.; Hochstrasser, M. Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol. 2008, 9, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Finley, D. Recognition and processing of ubiquitin-protein conjugates by the proteasome. Annu. Rev. Biochem. 2009, 78, 477–513. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, H.D.; Walden, H. Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell Biol. 2010, 11, 479–489. [Google Scholar] [CrossRef]
- Hirsch, C.; Gauss, R.; Horn, S.C.; Neuber, O.; Sommer, T. The ubiquitylation machinery of the endoplasmic reticulum. Nature 2009, 458, 453–460. [Google Scholar] [CrossRef]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef]
- Wertz, I.E.; Dixit, V.M. Regulation of death receptor signaling by the ubiquitin system. Cell Death Differ. 2010, 17, 14–24. [Google Scholar] [CrossRef]
- Kirkin, V.; McEwan, D.G.; Novak, I.; Dikic, I. A role for ubiquitin in selective autophagy. Mol. Cell 2009, 34, 259–269. [Google Scholar] [CrossRef]
- Hershko, A.; Ciechanover, A. The ubiquitin system. Annu. Rev. Biochem. 1998, 67, 425–479. [Google Scholar] [CrossRef]
- Clague, M.J.; Heride, C.; Urbe, S. The demographics of the ubiquitin system. Trends Cell Biol. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Barghout, S.H.; Schimmer, A.D. E1 Enzymes as Therapeutic Targets in Cancer. Pharmacol. Rev. 2021, 73, 1–58. [Google Scholar] [CrossRef] [PubMed]
- Mueller, D.L. E3 ubiquitin ligases as T cell anergy factors. Nat. Immunol. 2004, 5, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Kirisako, T.; Kamei, K.; Murata, S.; Kato, M.; Fukumoto, H.; Kanie, M.; Sano, S.; Tokunaga, F.; Tanaka, K.; Iwai, K. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006, 25, 4877–4887. [Google Scholar] [CrossRef]
- Husnjak, K.; Dikic, I. Ubiquitin-binding proteins: Decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem. 2012, 81, 291–322. [Google Scholar] [CrossRef]
- Herhaus, L.; Dikic, I. Expanding the ubiquitin code through post-translational modification. EMBO Rep. 2015, 16, 1071–1083. [Google Scholar] [CrossRef] [PubMed]
- Seeler, J.S.; Dejean, A. SUMO and the robustness of cancer. Nat. Rev. Cancer 2017, 17, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, K.D. DUBs at a glance. J. Cell Sci. 2009, 122, 2325–2329. [Google Scholar] [CrossRef]
- Komander, D.; Clague, M.J.; Urbe, S. Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009, 10, 550–563. [Google Scholar] [CrossRef]
- Clague, M.J.; Barsukov, I.; Coulson, J.M.; Liu, H.; Rigden, D.J.; Urbe, S. Deubiquitylases from genes to organism. Physiol. Rev. 2013, 93, 1289–1315. [Google Scholar] [CrossRef]
- Reyes-Turcu, F.E.; Ventii, K.H.; Wilkinson, K.D. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem. 2009, 78, 363–397. [Google Scholar] [CrossRef]
- Abdul Rehman, S.A.; Kristariyanto, Y.A.; Choi, S.Y.; Nkosi, P.J.; Weidlich, S.; Labib, K.; Hofmann, K.; Kulathu, Y. MINDY-1 Is a Member of an Evolutionarily Conserved and Structurally Distinct New Family of Deubiquitinating Enzymes. Mol. Cell 2016, 63, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Engilberge, S.; Girard, E.; Gabel, F.; Franzetti, B.; Maupin-Furlow, J.A. Structural Insight into Ubiquitin-Like Protein Recognition and Oligomeric States of JAMM/MPN+ Proteases. Structure 2017, 25, 823–833.e6. [Google Scholar] [CrossRef] [PubMed]
- Mevissen, T.E.T.; Komander, D. Mechanisms of Deubiquitinase Specificity and Regulation. Annu. Rev. Biochem. 2017, 86, 159–192. [Google Scholar] [CrossRef]
- Wilkinson, K.A.; Henley, J.M. Mechanisms, regulation and consequences of protein SUMOylation. Biochem. J. 2010, 428, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Gong, L.; Kamitani, T.; Wada, T.; Okura, I.; Wei, C.F.; Chang, H.M.; Yeh, E.T. Protection against Fas/APO-1- and tumor necrosis factor-mediated cell death by a novel protein, sentrin. J. Immunol. 1996, 157, 4277–4281. [Google Scholar] [CrossRef] [PubMed]
- Bohren, K.M.; Nadkarni, V.; Song, J.H.; Gabbay, K.H.; Owerbach, D. A M55V polymorphism in a novel SUMO gene (SUMO-4) differentially activates heat shock transcription factors and is associated with susceptibility to type I diabetes mellitus. J. Biol. Chem. 2004, 279, 27233–27238. [Google Scholar] [CrossRef]
- Mannen, H.; Tseng, H.M.; Cho, C.L.; Li, S.S. Cloning and expression of human homolog HSMT3 to yeast SMT3 suppressor of MIF2 mutations in a centromere protein gene. Biochem. Biophys. Res. Commun. 1996, 222, 178–180. [Google Scholar] [CrossRef]
- Lapenta, V.; Chiurazzi, P.; van der Spek, P.; Pizzuti, A.; Hanaoka, F.; Brahe, C. SMT3A, a human homologue of the S. cerevisiae SMT3 gene, maps to chromosome 21qter and defines a novel gene family. Genomics 1997, 40, 362–366. [Google Scholar] [CrossRef]
- Liang, Y.C.; Lee, C.C.; Yao, Y.L.; Lai, C.C.; Schmitz, M.L.; Yang, W.M. SUMO5, a Novel Poly-SUMO Isoform, Regulates PML Nuclear Bodies. Sci. Rep. 2016, 6, 26509. [Google Scholar] [CrossRef]
- Tatham, M.H.; Jaffray, E.; Vaughan, O.A.; Desterro, J.M.; Botting, C.H.; Naismith, J.H.; Hay, R.T. Polymeric chains of SUMO-2 and SUMO-3 are conjugated to protein substrates by SAE1/SAE2 and Ubc9. J. Biol. Chem. 2001, 276, 35368–35374. [Google Scholar] [CrossRef]
- Gareau, J.R.; Lima, C.D. The SUMO pathway: Emerging mechanisms that shape specificity, conjugation and recognition. Nat. Rev. Mol. Cell Biol. 2010, 11, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Yau, T.Y.; Sander, W.; Eidson, C.; Courey, A.J. SUMO Interacting Motifs: Structure and Function. Cells 2021, 10, 2825. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Salvesen, G.S. DeSUMOylating enzymes—SENPs. IUBMB Life 2008, 60, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Impens, F.; Radoshevich, L.; Cossart, P.; Ribet, D. Mapping of SUMO sites and analysis of SUMOylation changes induced by external stimuli. Proc. Natl. Acad. Sci. USA 2014, 111, 12432–12437. [Google Scholar] [CrossRef] [PubMed]
- Hay, R.T. SUMO: A history of modification. Mol. Cell 2005, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Durocher, D. Regulation of DNA damage responses by ubiquitin and SUMO. Mol. Cell 2013, 49, 795–807. [Google Scholar] [CrossRef]
- Muller, S.; Ledl, A.; Schmidt, D. SUMO: A regulator of gene expression and genome integrity. Oncogene 2004, 23, 1998–2008. [Google Scholar] [CrossRef]
- Eifler, K.; Vertegaal, A.C.O. SUMOylation-Mediated Regulation of Cell Cycle Progression and Cancer. Trends Biochem. Sci. 2015, 40, 779–793. [Google Scholar] [CrossRef]
- Stielow, B.; Sapetschnig, A.; Kruger, I.; Kunert, N.; Brehm, A.; Boutros, M.; Suske, G. Identification of SUMO-dependent chromatin-associated transcriptional repression components by a genome-wide RNAi screen. Mol. Cell 2008, 29, 742–754. [Google Scholar] [CrossRef]
- Terui, Y.; Saad, N.; Jia, S.; McKeon, F.; Yuan, J. Dual role of sumoylation in the nuclear localization and transcriptional activation of NFAT1. J. Biol. Chem. 2004, 279, 28257–28265. [Google Scholar] [CrossRef]
- Nathan, D.; Sterner, D.E.; Berger, S.L. Histone modifications: Now summoning sumoylation. Proc. Natl. Acad. Sci. USA 2003, 100, 13118–13120. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Giasson, B.I.; Glavis-Bloom, A.; Brewer, M.D.; Shorter, J.; Gitler, A.D.; Yang, X. A cellular system that degrades misfolded proteins and protects against neurodegeneration. Mol. Cell 2014, 55, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Kurka, T.; Jeggo, P.A. KAP-1 phosphorylation regulates CHD3 nucleosome remodeling during the DNA double-strand break response. Nat. Struct. Mol. Biol. 2011, 18, 831–839. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Mitchell, J.P.; Piguet, V. β-TrCP dependency of HIV-1 Vpu-induced downregulation of CD4 and BST-2/tetherin. Curr. HIV Res. 2012, 10, 307–314. [Google Scholar] [CrossRef]
- Estrabaud, E.; Le Rouzic, E.; Lopez-Vergès, S.; Morel, M.; Belaïdouni, N.; Benarous, R.; Transy, C.; Berlioz-Torrent, C.; Margottin-Goguet, F. Regulated degradation of the HIV-1 Vpu protein through a betaTrCP-independent pathway limits the release of viral particles. PLoS Pathog. 2007, 3, e104. [Google Scholar] [CrossRef]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A novel human WD protein, h-beta TrCp, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol. Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Jin, Y.J.; Cai, C.Y.; Zhang, X.; Burakoff, S.J. Lysine 144, a ubiquitin attachment site in HIV-1 Nef, is required for Nef-mediated CD4 down-regulation. J. Immunol. 2008, 180, 7878–7886. [Google Scholar] [CrossRef]
- daSilva, L.L.; Sougrat, R.; Burgos, P.V.; Janvier, K.; Mattera, R.; Bonifacino, J.S. Human immunodeficiency virus type 1 Nef protein targets CD4 to the multivesicular body pathway. J. Virol. 2009, 83, 6578–6590. [Google Scholar] [CrossRef]
- Madrid, R.; Janvier, K.; Hitchin, D.; Day, J.; Coleman, S.; Noviello, C.; Bouchet, J.; Benmerah, A.; Guatelli, J.; Benichou, S. Nef-induced alteration of the early/recycling endosomal compartment correlates with enhancement of HIV-1 infectivity. J. Biol. Chem. 2005, 280, 5032–5044. [Google Scholar] [CrossRef]
- Fernandis, A.Z.; Cherla, R.P.; Chernock, R.D.; Ganju, R.K. CXCR4/CCR5 down-modulation and chemotaxis are regulated by the proteasome pathway. J. Biol. Chem. 2002, 277, 18111–18117. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, M.; Ogata, Y.; Mahiti, M.; Maeda, Y.; Kuang, X.T.; Miura, T.; Jessen, H.; Walker, B.D.; Brockman, M.A.; Brumme, Z.L.; et al. Differential Ability of Primary HIV-1 Nef Isolates To Downregulate HIV-1 Entry Receptors. J. Virol. 2015, 89, 9639–9652. [Google Scholar] [CrossRef] [PubMed]
- Venzke, S.; Michel, N.; Allespach, I.; Fackler, O.T.; Keppler, O.T. Expression of Nef downregulates CXCR4, the major coreceptor of human immunodeficiency virus, from the surfaces of target cells and thereby enhances resistance to superinfection. J. Virol. 2006, 80, 11141–11152. [Google Scholar] [CrossRef] [PubMed]
- Nisole, S.; Stoye, J.P.; Saïb, A. TRIM family proteins: Retroviral restriction and antiviral defence. Nat. Rev. Microbiol. 2005, 3, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.J.; Towers, G.J. Inhibition of retroviral replication by members of the TRIM protein family. Curr. Top. Microbiol. Immunol. 2013, 371, 29–66. [Google Scholar]
- Sebastian, S.; Luban, J. TRIM5alpha selectively binds a restriction-sensitive retroviral capsid. Retrovirology 2005, 2, 40. [Google Scholar] [CrossRef]
- Stremlau, M.; Perron, M.; Lee, M.; Li, Y.; Song, B.; Javanbakht, H.; Diaz-Griffero, F.; Anderson, D.J.; Sundquist, W.I.; Sodroski, J. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. USA 2006, 103, 5514–5519. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef]
- Yamauchi, K.; Wada, K.; Tanji, K.; Tanaka, M.; Kamitani, T. Ubiquitination of E3 ubiquitin ligase TRIM5 alpha and its potential role. FEBS J. 2008, 275, 1540–1555. [Google Scholar] [CrossRef]
- Grütter, M.G.; Luban, J. TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2012, 2, 142–150. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Turelli, P.; Caron, G.; Friedli, M.; Perrin, L.; Trono, D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature 2003, 424, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yang, B.; Pomerantz, R.J.; Zhang, C.; Arunachalam, S.C.; Gao, L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature 2003, 424, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, E.S.; Yu, X.-F. Lentiviral Vif: Viral Hijacker of the Ubiquitin-Proteasome System. Int. J. Hematol. 2006, 83, 208–212. [Google Scholar] [CrossRef]
- Yu, X.; Yu, Y.; Liu, B.; Luo, K.; Kong, W.; Mao, P.; Yu, X.F. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science 2003, 302, 1056–1060. [Google Scholar] [CrossRef]
- Sugiyama, R.; Abe, M.; Nishitsuji, H.; Murakami, Y.; Takeuchi, H.; Takaku, H. Induction of heat-shock protein 70 by prostaglandin A1 inhibits HIV-1 Vif-mediated degradation of APOBEC3G. Antivir. Res. 2013, 99, 307–311. [Google Scholar] [CrossRef] [PubMed]
- Pan, T.; Song, Z.; Wu, L.; Liu, G.; Ma, X.; Peng, Z.; Zhou, M.; Liang, L.; Liu, B.; Liu, J.; et al. USP49 potently stabilizes APOBEC3G protein by removing ubiquitin and inhibits HIV-1 replication. eLife 2019, 8, e48318. [Google Scholar] [CrossRef]
- Stavrou, S.; Nitta, T.; Kotla, S.; Ha, D.; Nagashima, K.; Rein, A.R.; Fan, H.; Ross, S.R. Murine leukemia virus glycosylated Gag blocks apolipoprotein B editing complex 3 and cytosolic sensor access to the reverse transcription complex. Proc. Natl. Acad. Sci. USA 2013, 110, 9078–9083. [Google Scholar] [CrossRef]
- Berger, G.; Durand, S.; Fargier, G.; Nguyen, X.N.; Cordeil, S.; Bouaziz, S.; Muriaux, D.; Darlix, J.L.; Cimarelli, A. APOBEC3A is a specific inhibitor of the early phases of HIV-1 infection in myeloid cells. PLoS Pathog. 2011, 7, e1002221. [Google Scholar] [CrossRef]
- Romani, B.; Engelbrecht, S.; Glashoff, R.H. Antiviral roles of APOBEC proteins against HIV-1 and suppression by Vif. Arch. Virol. 2009, 154, 1579–1588. [Google Scholar] [CrossRef]
- Krisko, J.F.; Begum, N.; Baker, C.E.; Foster, J.L.; Garcia, J.V. APOBEC3G and APOBEC3F Act in Concert To Extinguish HIV-1 Replication. J. Virol. 2016, 90, 4681–4695. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Münk, C.; Schweizer, M.; Cichutek, K.; Schüle, S.; Flory, E. Interaction of Vpx and apolipoprotein B mRNA-editing catalytic polypeptide 3 family member A (APOBEC3A) correlates with efficient lentivirus infection of monocytes. J. Biol. Chem. 2010, 285, 12248–12254. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of HIV-1 infection of macrophages mediated by the SAMHD1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Wang, H.; Guo, H.; Su, J.; Rui, Y.; Zheng, W.; Gao, W.; Zhang, W.; Li, Z.; Liu, G.; Markham, R.B.; et al. Inhibition of Vpx-Mediated SAMHD1 and Vpr-Mediated Host Helicase Transcription Factor Degradation by Selective Disruption of Viral CRL4 (DCAF1) E3 Ubiquitin Ligase Assembly. J. Virol. 2017, 91, e00225-17. [Google Scholar] [CrossRef]
- Li, Z.; Huan, C.; Wang, H.; Liu, Y.; Liu, X.; Su, X.; Yu, J.; Zhao, Z.; Yu, X.-F.; Zheng, B.; et al. TRIM21-mediated proteasomal degradation of SAMHD1 regulates its antiviral activity. EMBO Rep. 2020, 21, e47528. [Google Scholar] [CrossRef]
- Mulder, L.C.; Muesing, M.A. Degradation of HIV-1 integrase by the N-end rule pathway. J. Biol. Chem. 2000, 275, 29749–29753. [Google Scholar] [CrossRef]
- Lloyd, A.G.; Ng, Y.S.; Muesing, M.A.; Simon, V.; Mulder, L.C. Characterization of HIV-1 integrase N-terminal mutant viruses. Virology 2007, 360, 129–135. [Google Scholar] [CrossRef]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef]
- Zheng, Y.; Ao, Z.; Wang, B.; Jayappa, K.D.; Yao, X. Host protein Ku70 binds and protects HIV-1 integrase from proteasomal degradation and is required for HIV replication. J. Biol. Chem. 2011, 286, 17722–17735. [Google Scholar] [CrossRef] [PubMed]
- Martin-Serrano, J. The role of ubiquitin in retroviral egress. Traffic 2007, 8, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Chau, V.; Wills, J.W. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. USA 2000, 97, 13069–13074. [Google Scholar] [CrossRef] [PubMed]
- Sette, P.; Nagashima, K.; Piper, R.C.; Bouamr, F. Ubiquitin conjugation to Gag is essential for ESCRT-mediated HIV-1 budding. Retrovirology 2013, 10, 79. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Ott, D.E.; Chertova, E.N.; Welker, R.; Tessmer, U.; Princiotta, M.F.; Bennink, J.R.; Krausslich, H.G.; Yewdell, J.W. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. USA 2000, 97, 13057–13062. [Google Scholar] [CrossRef]
- Ott, D.E.; Coren, L.V.; Copeland, T.D.; Kane, B.P.; Johnson, D.G.; Sowder, R.C.; Yoshinaka, Y.; Oroszlan, S.; Arthur, L.O.; Henderson, L.E. Ubiquitin Is Covalently Attached to the p6Gag Proteins of Human Immunodeficiency Virus Type 1 and Simian Immunodeficiency Virus and to the p12Gag Protein of Moloney Murine Leukemia Virus. J. Virol. 1998, 72, 2962–2968. [Google Scholar] [CrossRef]
- Demirov, D.G.; Freed, E.O. Retrovirus budding. Virus Res. 2004, 106, 87–102. [Google Scholar] [CrossRef]
- Welker, L.; Paillart, J.C.; Bernacchi, S. Importance of Viral Late Domains in Budding and Release of Enveloped RNA Viruses. Viruses 2021, 13, 1559. [Google Scholar] [CrossRef]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Côté, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Strack, B.; Calistri, A.; Craig, S.; Popova, E.; Göttlinger, H.G. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell 2003, 114, 689–699. [Google Scholar] [CrossRef]
- Strickland, M.; Ehrlich, L.S.; Watanabe, S.; Khan, M.; Strub, M.P.; Luan, C.H.; Powell, M.D.; Leis, J.; Tjandra, N.; Carter, C.A. Tsg101 chaperone function revealed by HIV-1 assembly inhibitors. Nat. Commun. 2017, 8, 1391. [Google Scholar] [CrossRef] [PubMed]
- Goila-Gaur, R.; Demirov, D.G.; Orenstein, J.M.; Ono, A.; Freed, E.O. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J. Virol. 2003, 77, 6507–6519. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Morita, E.; von Schwedler, U.; Müller, B.; Kräusslich, H.G.; Sundquist, W.I. NEDD4L overexpression rescues the release and infectivity of human immunodeficiency virus type 1 constructs lacking PTAP and YPXL late domains. J. Virol. 2008, 82, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Kikonyogo, A.; Bouamr, F.; Vana, M.L.; Xiang, Y.; Aiyar, A.; Carter, C.; Leis, J. Proteins related to the Nedd4 family of ubiquitin protein ligases interact with the L domain of Rous sarcoma virus and are required for gag budding from cells. Proc. Natl. Acad. Sci. USA 2001, 98, 11199–11204. [Google Scholar] [CrossRef] [PubMed]
- Blot, V.; Perugi, F.; Gay, B.; Prévost, M.C.; Briant, L.; Tangy, F.; Abriel, H.; Staub, O.; Dokhélar, M.C.; Pique, C. Nedd4.1-mediated ubiquitination and subsequent recruitment of Tsg101 ensure HTLV-1 Gag trafficking towards the multivesicular body pathway prior to virus budding. J. Cell Sci. 2004, 117, 2357–2367. [Google Scholar] [CrossRef]
- Jouvenet, N.; Neil, S.J.; Zhadina, M.; Zang, T.; Kratovac, Z.; Lee, Y.; McNatt, M.; Hatziioannou, T.; Bieniasz, P.D. Broad-spectrum inhibition of retroviral and filoviral particle release by tetherin. J. Virol. 2009, 83, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Tokarev, A.A.; Munguia, J.; Guatelli, J.C. Serine-threonine ubiquitination mediates downregulation of BST-2/tetherin and relief of restricted virion release by HIV-1 Vpu. J. Virol. 2011, 85, 51–63. [Google Scholar] [CrossRef]
- Janvier, K.; Pelchen-Matthews, A.; Renaud, J.B.; Caillet, M.; Marsh, M.; Berlioz-Torrent, C. The ESCRT-0 component HRS is required for HIV-1 Vpu-mediated BST-2/tetherin down-regulation. PLoS Pathog. 2011, 7, e1001265. [Google Scholar] [CrossRef]
- Zhang, F.; Landford, W.N.; Ng, M.; McNatt, M.W.; Bieniasz, P.D.; Hatziioannou, T. SIV Nef Proteins Recruit the AP-2 Complex to Antagonize Tetherin and Facilitate Virion Release. PLoS Pathog. 2011, 7, e1002039. [Google Scholar] [CrossRef]
- Hauser, H.; Lopez, L.A.; Yang, S.J.; Oldenburg, J.E.; Exline, C.M.; Guatelli, J.C.; Cannon, P.M. HIV-1 Vpu and HIV-2 Env counteract BST-2/tetherin by sequestration in a perinuclear compartment. Retrovirology 2010, 7, 51. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Mlcochova, P.; Pelchen-Matthews, A.; Petit, S.J.; Mattiuzzo, G.; Pillay, D.; Takeuchi, Y.; Marsh, M.; Towers, G.J. Simian immunodeficiency virus envelope glycoprotein counteracts tetherin/BST-2/CD317 by intracellular sequestration. Proc. Natl. Acad. Sci. USA 2009, 106, 20889–20894. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Pacini, G.; Berlioz-Torrent, C.; Janvier, K. Characterization of E3 ligases involved in lysosomal sorting of the HIV-1 restriction factor BST2. J. Cell Sci. 2017, 130, 1596–1611. [Google Scholar] [CrossRef] [PubMed]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.-F.; Matsuda, Z.; Yu, Q.-C.; Lee, T.-H.; Essex, M. Role of the C terminus Gag protein in human immunodeficiency virus type 1 virion assembly and maturation. J. Gen. Virol. 1995, 76, 3171–3179. [Google Scholar] [CrossRef] [PubMed]
- Solbak, S.M.Ø.; Reksten, T.R.; Hahn, F.; Wray, V.; Henklein, P.; Henklein, P.; Halskau, Ø.; Schubert, U.; Fossen, T. HIV-1 p6—A structured to flexible multifunctional membrane-interacting protein. Biochim. Biophys. Acta (BBA)—Biomembr. 2013, 1828, 816–823. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, E.; Kräusslich, H.-G. Analysis of Human Immunodeficiency Virus Type 1 Gag Ubiquitination. J. Virol. 2005, 79, 9134–9144. [Google Scholar] [CrossRef]
- Gurer, C.; Berthoux, L.; Luban, J. Covalent Modification of Human Immunodeficiency Virus Type 1 p6 by SUMO-1. J. Virol. 2005, 79, 910–917. [Google Scholar] [CrossRef]
- Carlton, J.G.; Martin-Serrano, J. Parallels Between Cytokinesis and Retroviral Budding: A Role for the ESCRT Machinery. Science 2007, 316, 1908–1912. [Google Scholar] [CrossRef]
- Terreni, M.; Valentini, P.; Liverani, V.; Gutierrez, M.I.; Di Primio, C.; Di Fenza, A.; Tozzini, V.; Allouch, A.; Albanese, A.; Giacca, M.; et al. GCN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 2010, 7, 18. [Google Scholar] [CrossRef]
- Topper, M.; Luo, Y.; Zhadina, M.; Mohammed, K.; Smith, L.; Muesing, M.A. Posttranslational Acetylation of the Human Immunodeficiency Virus Type 1 Integrase Carboxyl-Terminal Domain Is Dispensable for Viral Replication. J. Virol. 2007, 81, 3012–3017. [Google Scholar] [CrossRef] [PubMed]
- Okino, Y.; Inayoshi, Y.; Kojima, Y.; Kidani, S.; Kaneoka, H.; Honkawa, A.; Higuchi, H.; Nishijima, K.-i.; Miyake, K.; Iijima, S. Moloney murine leukemia virus integrase and reverse transcriptase interact with PML proteins. J. Biochem. 2012, 152, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Zamborlini, A.; Coiffic, A.; Beauclair, G.; Delelis, O.; Paris, J.; Koh, Y.; Magne, F.; Giron, M.-L.; Tobaly-Tapiero, J.; Deprez, E.; et al. Impairment of Human Immunodeficiency Virus Type-1 Integrase SUMOylation Correlates with an Early Replication Defect. J. Biol. Chem. 2011, 286, 21013–21022. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Yao, X. Posttranslational Modifications of HIV-1 Integrase by Various Cellular Proteins during Viral Replication. Viruses 2013, 5, 1787–1801. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Jayappa, K.D.; Ao, Z.; Qiu, X.; Su, R.-C.; Yao, X. Noncovalent SUMO-interaction motifs in HIV integrase play important roles in SUMOylation, cofactor binding, and virus replication. Virol. J. 2019, 16, 42. [Google Scholar] [CrossRef] [PubMed]
- Alin, K.; Goff, S.P. Amino Acid Substitutions in the CA Protein of Moloney Murine Leukemia Virus That Block Early Events in Infection. Virology 1996, 222, 339–351. [Google Scholar] [CrossRef]
- Yueh, A.; Leung, J.; Bhattacharyya, S.; Perrone, L.A.; Santos, K.D.L.; Pu, S.-Y.; Goff, S.P. Interaction of Moloney Murine Leukemia Virus Capsid with Ubc9 and PIASy Mediates SUMO-1 Addition Required Early in Infection. J. Virol. 2006, 80, 342–352. [Google Scholar] [CrossRef]
- Kehn, K.; Berro, R.; de la Fuente, C.; Strouss, K.; Ghedin, E.; Dadgar, S.; Bottazzi, M.E.; Pumfery, A.; Kashanchi, F. Mechanisms of HTLV-1 transformation. Front. Biosci. 2004, 9, 2347–2372. [Google Scholar] [CrossRef]
- Lamsoul, I.; Lodewick, J.; Lebrun, S.; Brasseur, R.; Burny, A.; Gaynor, R.B.; Bex, F. Exclusive ubiquitination and sumoylation on overlapping lysine residues mediate NF-kappaB activation by the human T-cell leukemia virus tax oncoprotein. Mol. Cell Biol. 2005, 25, 10391–10406. [Google Scholar] [CrossRef]
- Turci, M.; Lodewick, J.; Di Gennaro, G.; Rinaldi, A.S.; Marin, O.; Diani, E.; Sampaio, C.; Bex, F.; Bertazzoni, U.; Romanelli, M.G. Ubiquitination and sumoylation of the HTLV-2 Tax-2B protein regulate its NF-kappaB activity: A comparative study with the HTLV-1 Tax-1 protein. Retrovirology 2012, 9, 102. [Google Scholar] [CrossRef]
- Turci, M.; Lodewick, J.; Righi, P.; Polania, A.; Romanelli, M.G.; Bex, F.; Bertazzoni, U. HTLV-2B Tax oncoprotein is modified by ubiquitination and sumoylation and displays intracellular localization similar to its homologue HTLV-1 Tax. Virology 2009, 386, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Fryrear, K.A.; Guo, X.; Kerscher, O.; Semmes, O.J. The Sumo-targeted ubiquitin ligase RNF4 regulates the localization and function of the HTLV-1 oncoprotein Tax. Blood 2012, 119, 1173–1181. [Google Scholar] [CrossRef] [PubMed]
- Pene, S.; Waast, L.; Bonnet, A.; Benit, L.; Pique, C. A non-SUMOylated tax protein is still functional for NF-kappaB pathway activation. J. Virol. 2014, 88, 10655–10661. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, T.; Singh, D.P.; Fatma, N. LEDGF, a survival factor, activates stress-related genes. Prog. Retin. Eye Res. 2002, 21, 341–358. [Google Scholar] [CrossRef]
- Turlure, F.; Maertens, G.; Rahman, S.; Cherepanov, P.; Engelman, A. A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo. Nucleic Acids Res. 2006, 34, 1653–1665. [Google Scholar] [CrossRef]
- Cherepanov, P.; Devroe, E.; Silver, P.A.; Engelman, A. Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase. J. Biol. Chem. 2004, 279, 48883–48892. [Google Scholar] [CrossRef]
- Shun, M.C.; Raghavendra, N.K.; Vandegraaff, N.; Daigle, J.E.; Hughes, S.; Kellam, P.; Cherepanov, P.; Engelman, A. LEDGF/p75 functions downstream from preintegration complex formation to effect gene-specific HIV-1 integration. Genes Dev. 2007, 21, 1767–1778. [Google Scholar] [CrossRef]
- Bueno, M.T.; Garcia-Rivera, J.A.; Kugelman, J.R.; Morales, E.; Rosas-Acosta, G.; Llano, M. SUMOylation of the lens epithelium-derived growth factor/p75 attenuates its transcriptional activity on the heat shock protein 27 promoter. J. Mol. Biol. 2010, 399, 221–239. [Google Scholar] [CrossRef]
- DeLuca, C.; Petropoulos, L.; Zmeureanu, D.; Hiscott, J. Nuclear IkappaBbeta maintains persistent NF-kappaB activation in HIV-1-infected myeloid cells. J. Biol. Chem. 1999, 274, 13010–13016. [Google Scholar] [CrossRef]
- Hay, R.T.; Vuillard, L.; Desterro, J.M.; Rodriguez, M.S. Control of NF-kappa B transcriptional activation by signal induced proteolysis of I kappa B alpha. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999, 354, 1601–1609. [Google Scholar] [CrossRef]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF-kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Romanchikova, N.; Ivanova, V.; Scheller, C.; Jankevics, E.; Jassoy, C.; Serfling, E. NFAT transcription factors control HIV-1 expression through a binding site downstream of TAR region. Immunobiology 2003, 208, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Glockner-Pagel, J.; Vaeth, M.; Schumann, J.E.; Buttmann, M.; Bopp, T.; Schmitt, E.; Serfling, E.; Berberich-Siebelt, F. Sumoylation of the transcription factor NFATc1 leads to its subnuclear relocalization and interleukin-2 repression by histone deacetylase. J. Biol. Chem. 2009, 284, 10935–10946. [Google Scholar] [CrossRef] [PubMed]
- Groh, S.; Schotta, G. Silencing of endogenous retroviruses by heterochromatin. Cell. Mol. Life Sci. 2017, 74, 2055–2065. [Google Scholar] [CrossRef]
- Yang, B.X.; El Farran, C.A.; Guo, H.C.; Yu, T.; Fang, H.T.; Wang, H.F.; Schlesinger, S.; Seah, Y.F.; Goh, G.Y.; Neo, S.P.; et al. Systematic identification of factors for provirus silencing in embryonic stem cells. Cell 2015, 163, 230–245. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.J.; Dulberg, V.; Moon, K.M.; Foster, L.J.; Chen, C.; Karimi, M.M.; Lorincz, M.C. hnRNP K coordinates transcriptional silencing by SETDB1 in embryonic stem cells. PLoS Genet. 2015, 11, e1004933. [Google Scholar] [CrossRef]
- Groh, S.; Milton, A.V.; Marinelli, L.K.; Sickinger, C.V.; Russo, A.; Bollig, H.; de Almeida, G.P.; Schmidt, A.; Forne, I.; Imhof, A.; et al. Morc3 silences endogenous retroviruses by enabling Daxx-mediated histone H3.3 incorporation. Nat. Commun. 2021, 12, 5996. [Google Scholar] [CrossRef]
- Zhang, L.; Qin, J.; Li, Y.; Wang, J.; He, Q.; Zhou, J.; Liu, M.; Li, D. Modulation of the stability and activities of HIV-1 Tat by its ubiquitination and carboxyl-terminal region. Cell Biosci. 2014, 4, 61. [Google Scholar] [CrossRef]
- Lata, S.; Ali, A.; Sood, V.; Raja, R.; Banerjea, A.C. HIV-1 Rev downregulates Tat expression and viral replication via modulation of NAD(P)H:quinine oxidoreductase 1 (NQO1). Nat. Commun. 2015, 6, 7244. [Google Scholar] [CrossRef]
- Ali, A.; Raja, R.; Farooqui, S.R.; Ahmad, S.; Banerjea, A.C. USP7 deubiquitinase controls HIV-1 production by stabilizing Tat protein. Biochem. J. 2017, 474, 1653–1668. [Google Scholar] [CrossRef]
- Brès, V.; Kiernan, R.E.; Linares, L.K.; Chable-Bessia, C.; Plechakova, O.; Tréand, C.; Emiliani, S.; Peloponese, J.M.; Jeang, K.T.; Coux, O.; et al. A non-proteolytic role for ubiquitin in Tat-mediated transactivation of the HIV-1 promoter. Nat. Cell Biol. 2003, 5, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Farooqui, S.R.; Banerjea, A.C. The host cell ubiquitin ligase protein CHIP is a potent suppressor of HIV-1 replication. J. Biol. Chem. 2019, 294, 7283–7295. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Li, G.; Zhao, S.; Wang, H.; Huan, C.; Zheng, B.; Jiang, C.; Zhang, W. Deubiquitinating Enzyme USP21 Inhibits HIV-1 Replication by Downregulating Tat Expression. J. Virol. 2021, 95, e0046021. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, R.; Naganuma, H.; Nishitsuji, H.; Takaku, H. Human immunodeficiency virus-1 Nef suppresses Hsp70-mediated Tat activation. FEBS Lett. 2011, 585, 3367–3371. [Google Scholar] [CrossRef]
- Hong, H.W.; Lee, S.W.; Myung, H. Induced degradation of Tat by nucleocapsid (NC) via the proteasome pathway and its effect on HIV transcription. Viruses 2013, 5, 1143–1152. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Zhang, Q.; Wu, Z.; Chen, P.; Huang, Y.; Liu, S.; Li, L. UHRF1 Suppresses HIV-1 Transcription and Promotes HIV-1 Latency by Competing with p-TEFb for Ubiquitination-Proteasomal Degradation of Tat. mBio 2021, 12, e0162521. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell. Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Kim, J.B.; Sharp, P.A. Positive transcription elongation factor B phosphorylates hSPT5 and RNA polymerase II carboxyl-terminal domain independently of cyclin-dependent kinase-activating kinase. J. Biol. Chem. 2001, 276, 12317–12323. [Google Scholar] [CrossRef]
- Li, J.; Liu, Y.; Rhee, H.S.; Ghosh, S.K.; Bai, L.; Pugh, B.F.; Gilmour, D.S. Kinetic competition between elongation rate and binding of NELF controls promoter-proximal pausing. Mol. Cell 2013, 50, 711–722. [Google Scholar] [CrossRef]
- Schulze-Gahmen, U.; Hurley, J.H. Structural mechanism for HIV-1 TAR loop recognition by Tat and the super elongation complex. Proc. Natl. Acad. Sci. USA 2018, 115, 12973–12978. [Google Scholar] [CrossRef]
- Liu, M.; Hsu, J.; Chan, C.; Li, Z.; Zhou, Q. The ubiquitin ligase Siah1 controls ELL2 stability and formation of super elongation complexes to modulate gene transcription. Mol. Cell 2012, 46, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Ruelas, D.S.; Chan, J.K.; Oh, E.; Heidersbach, A.J.; Hebbeler, A.M.; Chavez, L.; Verdin, E.; Rape, M.; Greene, W.C. MicroRNA-155 Reinforces HIV Latency. J. Biol. Chem. 2015, 290, 13736–13748. [Google Scholar] [CrossRef] [PubMed]
- Kashanchi, F.; Brady, J.N. Transcriptional and post-transcriptional gene regulation of HTLV-1. Oncogene 2005, 24, 5938–5951. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Yamamoto, M.; Akira, S.; Harhaj, E.W. The human T-cell leukemia virus type 1 Tax oncoprotein requires the ubiquitin-conjugating enzyme Ubc13 for NF-kappaB activation. J. Virol. 2007, 81, 13735–13742. [Google Scholar] [CrossRef] [PubMed]
- Yan, P.; Fu, J.; Qu, Z.; Li, S.; Tanaka, T.; Grusby, M.J.; Xiao, G. PDLIM2 suppresses human T-cell leukemia virus type I Tax-mediated tumorigenesis by targeting Tax into the nuclear matrix for proteasomal degradation. Blood 2009, 113, 4370–4380. [Google Scholar] [CrossRef]
- Andresen, V.; Pise-Masison, C.A.; Sinha-Datta, U.; Bellon, M.; Valeri, V.; Washington Parks, R.; Cecchinato, V.; Fukumoto, R.; Nicot, C.; Franchini, G. Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles. Blood 2011, 118, 1549–1559. [Google Scholar] [CrossRef]
- Yamaoka, S.; Courtois, G.; Bessia, C.; Whiteside, S.T.; Weil, R.; Agou, F.; Kirk, H.E.; Kay, R.J.; Israël, A. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell 1998, 93, 1231–1240. [Google Scholar] [CrossRef]
- Kumar, A.; Darcis, G.; Van Lint, C.; Herbein, G. Epigenetic control of HIV-1 post integration latency: Implications for therapy. Clin. Epigenet. 2015, 7, 103. [Google Scholar] [CrossRef]
- Imai, K.; Ochiai, K. Role of histone modification on transcriptional regulation and HIV-1 gene expression: Possible mechanisms of periodontal diseases in AIDS progression. J. Oral Sci. 2011, 53, 1–13. [Google Scholar] [CrossRef]
- Kulkarni, A.; Taylor, G.P.; Klose, R.J.; Schofield, C.J.; Bangham, C.R. Histone H2A monoubiquitylation and p38-MAPKs regulate immediate-early gene-like reactivation of latent retrovirus HTLV-1. JCI Insight 2018, 3, e123196. [Google Scholar] [CrossRef]
- Ping, Y.H.; Rana, T.M. DSIF and NELF interact with RNA polymerase II elongation complex and HIV-1 Tat stimulates P-TEFb-mediated phosphorylation of RNA polymerase II and DSIF during transcription elongation. J. Biol. Chem. 2001, 276, 12951–12958. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Geyer, M.; Zhou, Q. The control of HIV transcription: Keeping RNA polymerase II on track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Yang, T.; Luo, Y.; Wu, L.; Jiang, Y.; Song, Z.; Pan, T.; Liu, B.; Liu, G.; Liu, J.; et al. TRIM28 promotes HIV-1 latency by SUMOylating CDK9 and inhibiting P-TEFb. eLife 2019, 8, e42426. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Chen, T.; Peng, Z.; Wang, Z.; Liu, J.; Yang, T.; Wu, L.; Liu, G.; Zhou, M.; Tong, M.; et al. Histone chaperone CAF-1 promotes HIV-1 latency by leading the formation of phase-separated suppressive nuclear bodies. EMBO J. 2021, 40, e106632. [Google Scholar] [CrossRef] [PubMed]
- Zitouni, S.; Nabais, C.; Jana, S.C.; Guerrero, A.; Bettencourt-Dias, M. Polo-like kinases: Structural variations lead to multiple functions. Nat. Rev. Mol. Cell Biol. 2014, 15, 433–452. [Google Scholar] [CrossRef] [PubMed]
- Colnaghi, R.; Wheatley, S.P. Liaisons between survivin and Plk1 during cell division and cell death. J. Biol. Chem. 2010, 285, 22592–22604. [Google Scholar] [CrossRef]
- Liu, X.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef]
- Wen, D.; Wu, J.; Wang, L.; Fu, Z. SUMOylation Promotes Nuclear Import and Stabilization of Polo-like Kinase 1 to Support Its Mitotic Function. Cell Rep. 2017, 21, 2147–2159. [Google Scholar] [CrossRef]
- Zhou, D.; Hayashi, T.; Jean, M.; Kong, W.; Fiches, G.; Biswas, A.; Liu, S.; Yosief, H.O.; Zhang, X.; Bradner, J.; et al. Inhibition of Polo-like kinase 1 (PLK1) facilitates the elimination of HIV-1 viral reservoirs in CD4+ T cells ex vivo. Sci. Adv. 2020, 6, eaba1941. [Google Scholar] [CrossRef]
- Bosque, A.; Nilson, K.A.; Macedo, A.B.; Spivak, A.M.; Archin, N.M.; Van Wagoner, R.M.; Martins, L.J.; Novis, C.L.; Szaniawski, M.A.; Ireland, C.M.; et al. Benzotriazoles Reactivate Latent HIV-1 through Inactivation of STAT5 SUMOylation. Cell Rep. 2017, 18, 1324–1334. [Google Scholar] [CrossRef]
- Van Nguyen, T.; Angkasekwinai, P.; Dou, H.; Lin, F.M.; Lu, L.S.; Cheng, J.; Chin, Y.E.; Dong, C.; Yeh, E.T. SUMO-specific protease 1 is critical for early lymphoid development through regulation of STAT5 activation. Mol. Cell 2012, 45, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Miyakawa, K.; Ryo, A.; Murakami, T.; Ohba, K.; Yamaoka, S.; Fukuda, M.; Guatelli, J.; Yamamoto, N. BCA2/Rabring7 Promotes Tetherin-Dependent HIV-1 Restriction. PLoS Pathog. 2009, 5, e1000700. [Google Scholar] [CrossRef] [PubMed]
- Nityanandam, R.; Serra-Moreno, R. BCA2/Rabring7 Targets HIV-1 Gag for Lysosomal Degradation in a Tetherin-Independent Manner. PLoS Pathog. 2014, 10, e1004151. [Google Scholar] [CrossRef] [PubMed]
- Colomer-Lluch, M.; Serra-Moreno, R. BCA2/Rabring7 Interferes with HIV-1 Proviral Transcription by Enhancing the SUMOylation of IκBα. J. Virol. 2017, 91, e02098-16. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Lucic, B.; Forcato, M.; Penzo, C.; Billingsley, J.; Laketa, V.; Bosinger, S.; Stanic, M.; Gregoretti, F.; Antonelli, L.; et al. Alterations of redox and iron metabolism accompany the development of HIV latency. EMBO J. 2020, 39, e102209. [Google Scholar] [CrossRef]
- Aragon, L. The Smc5/6 Complex: New and Old Functions of the Enigmatic Long-Distance Relative. Annu. Rev. Genet. 2018, 52, 89–107. [Google Scholar] [CrossRef]
- Wu, L.; Pan, T.; Zhou, M.; Chen, T.; Wu, S.; Lv, X.; Liu, J.; Yu, F.; Guan, Y.; Liu, B.; et al. CBX4 contributes to HIV-1 latency by forming phase-separated nuclear bodies and SUMOylating EZH2. EMBO Rep. 2022, 23, e53855. [Google Scholar] [CrossRef]
- Lusic, M.; Marini, B.; Ali, H.; Lucic, B.; Luzzati, R.; Giacca, M. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host Microbe 2013, 13, 665–677. [Google Scholar] [CrossRef]
- Banani, S.F.; Rice, A.M.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663. [Google Scholar] [CrossRef]
- Corpet, A.; Kleijwegt, C.; Roubille, S.; Juillard, F.; Jacquet, K.; Texier, P.; Lomonte, P. PML nuclear bodies and chromatin dynamics: Catch me if you can! Nucleic Acids Res. 2020, 48, 11890–11912. [Google Scholar] [CrossRef]
- Andrews, E.A.; Palecek, J.; Sergeant, J.; Taylor, E.; Lehmann, A.R.; Watts, F.Z. Nse2, a component of the Smc5-6 complex, is a SUMO ligase required for the response to DNA damage. Mol. Cell Biol. 2005, 25, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Varejao, N.; Ibars, E.; Lascorz, J.; Colomina, N.; Torres-Rosell, J.; Reverter, D. DNA activates the Nse2/Mms21 SUMO E3 ligase in the Smc5/6 complex. EMBO J. 2018, 37, e98306. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.Y.; Hochstrasser, M. Histone sumoylation and chromatin dynamics. Nucleic Acids Res. 2021, 49, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Shiio, Y.; Eisenman, R.N. Histone sumoylation is associated with transcriptional repression. Proc. Natl. Acad. Sci. USA 2003, 100, 13225–13230. [Google Scholar] [CrossRef]
- Irwan, I.D.; Bogerd, H.P.; Cullen, B.R. Epigenetic silencing by the SMC5/6 complex mediates HIV-1 latency. Nat. Microbiol. 2022, 7, 2101–2113. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Schambelan, M.; Benson, C.A.; Carr, A.; Currier, J.S.; Dubé, M.P.; Gerber, J.G.; Grinspoon, S.K.; Grunfeld, C.; Kotler, D.P.; Mulligan, K.; et al. Management of Metabolic Complications Associated With Antiretroviral Therapy for HIV-1 Infection: Recommendations of an International AIDS Society-USA Panel. J. Acquir. Immune Defic. Syndr. 2002, 31, 257–275. [Google Scholar] [CrossRef]
- Barton, K.M.; Burch, B.D.; Soriano-Sarabia, N.; Margolis, D.M. Prospects for treatment of latent HIV. Clin. Pharm. 2013, 93, 46–56. [Google Scholar] [CrossRef]
- Madu, I.G.; Li, S.; Li, B.; Li, H.; Chang, T.; Li, Y.J.; Vega, R.; Rossi, J.; Yee, J.K.; Zaia, J.; et al. A Novel Class of HIV-1 Antiviral Agents Targeting HIV via a SUMOylation-Dependent Mechanism. Sci. Rep. 2015, 5, 17808. [Google Scholar] [CrossRef]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; de The, H.; Bazarbachi, A. ATL response to arsenic/interferon therapy is triggered by SUMO/PML/RNF4-dependent Tax degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.; Iketani, S.; Wang, P.; Jia, M.; Sahi, V.; Ho, D.D. CRISPR-based gene knockout screens reveal deubiquitinases involved in HIV-1 latency in two Jurkat cell models. Sci. Rep. 2020, 10, 5350. [Google Scholar] [CrossRef] [PubMed]
- Bobardt, M.; Kuo, J.; Chatterji, U.; Chanda, S.; Little, S.J.; Wiedemann, N.; Vuagniaux, G.; Gallay, P.A. The inhibitor apoptosis protein antagonist Debio 1143 Is an attractive HIV-1 latency reversal candidate. PLoS ONE 2019, 14, e0211746. [Google Scholar] [CrossRef]
- Kroonen, J.S.; Vertegaal, A.C.O. Targeting SUMO Signaling to Wrestle Cancer. Trends Cancer 2021, 7, 496–510. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, T.; Li, G.; Lu, Y.; Hu, M.; Ma, X. The Involvement of Ubiquitination and SUMOylation in Retroviruses Infection and Latency. Viruses 2023, 15, 985. https://doi.org/10.3390/v15040985
Liang T, Li G, Lu Y, Hu M, Ma X. The Involvement of Ubiquitination and SUMOylation in Retroviruses Infection and Latency. Viruses. 2023; 15(4):985. https://doi.org/10.3390/v15040985
Chicago/Turabian StyleLiang, Taizhen, Guojie Li, Yunfei Lu, Meilin Hu, and Xiancai Ma. 2023. "The Involvement of Ubiquitination and SUMOylation in Retroviruses Infection and Latency" Viruses 15, no. 4: 985. https://doi.org/10.3390/v15040985