

Conserved Sequences in the 5′ and 3′ Untranslated Regions of Jingmenvirus Group Representatives
 , and
, and 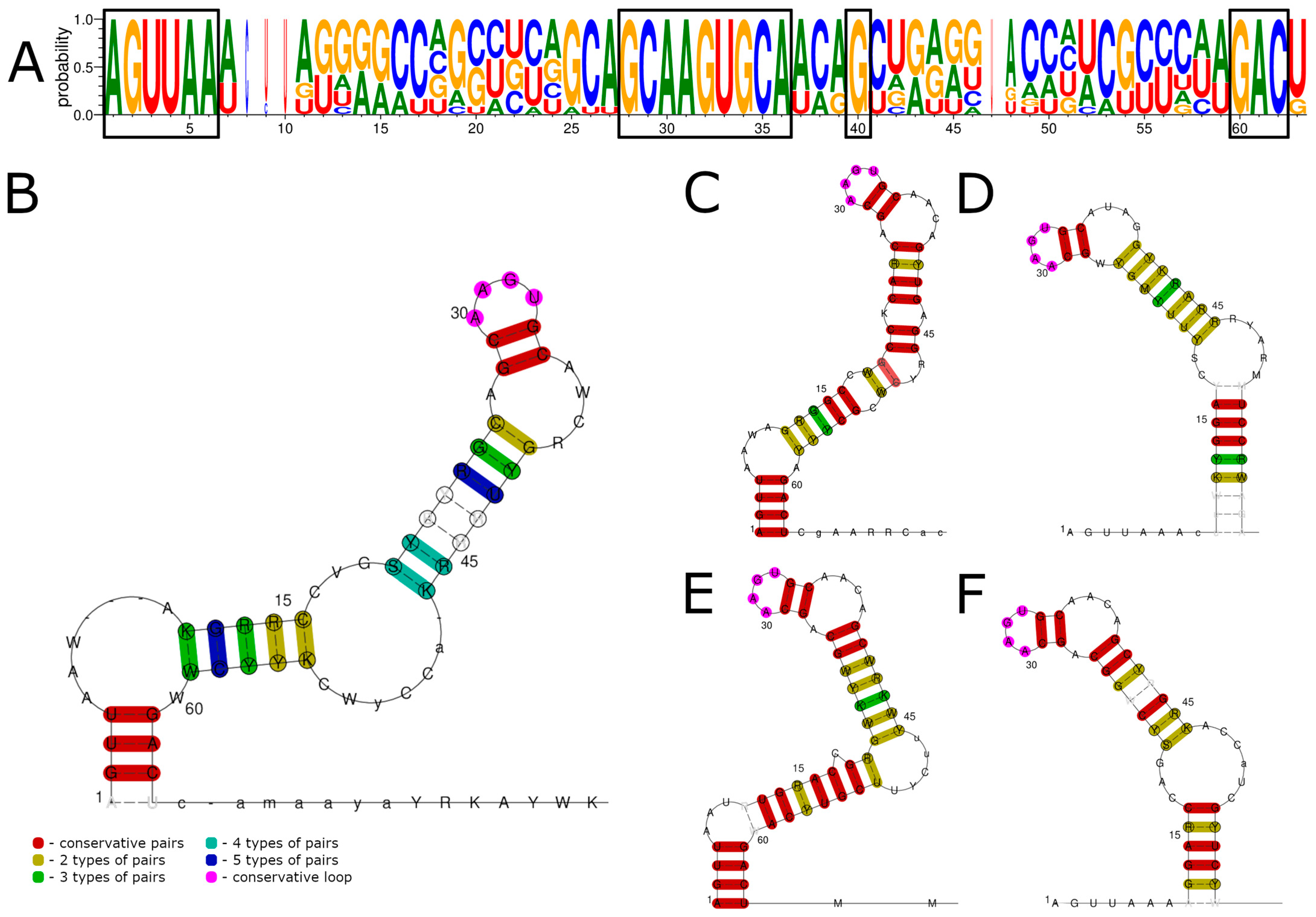{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cell Cultures
2.2. Obtaining of Virus-Containing Material
2.3. Rapid Amplification of cDNA Ends
2.4. Cloning of PCR Products
2.5. Sequencing
2.6. GenBank Dataset Preparation
2.7. Bioinformatics and Visualization
3. Results
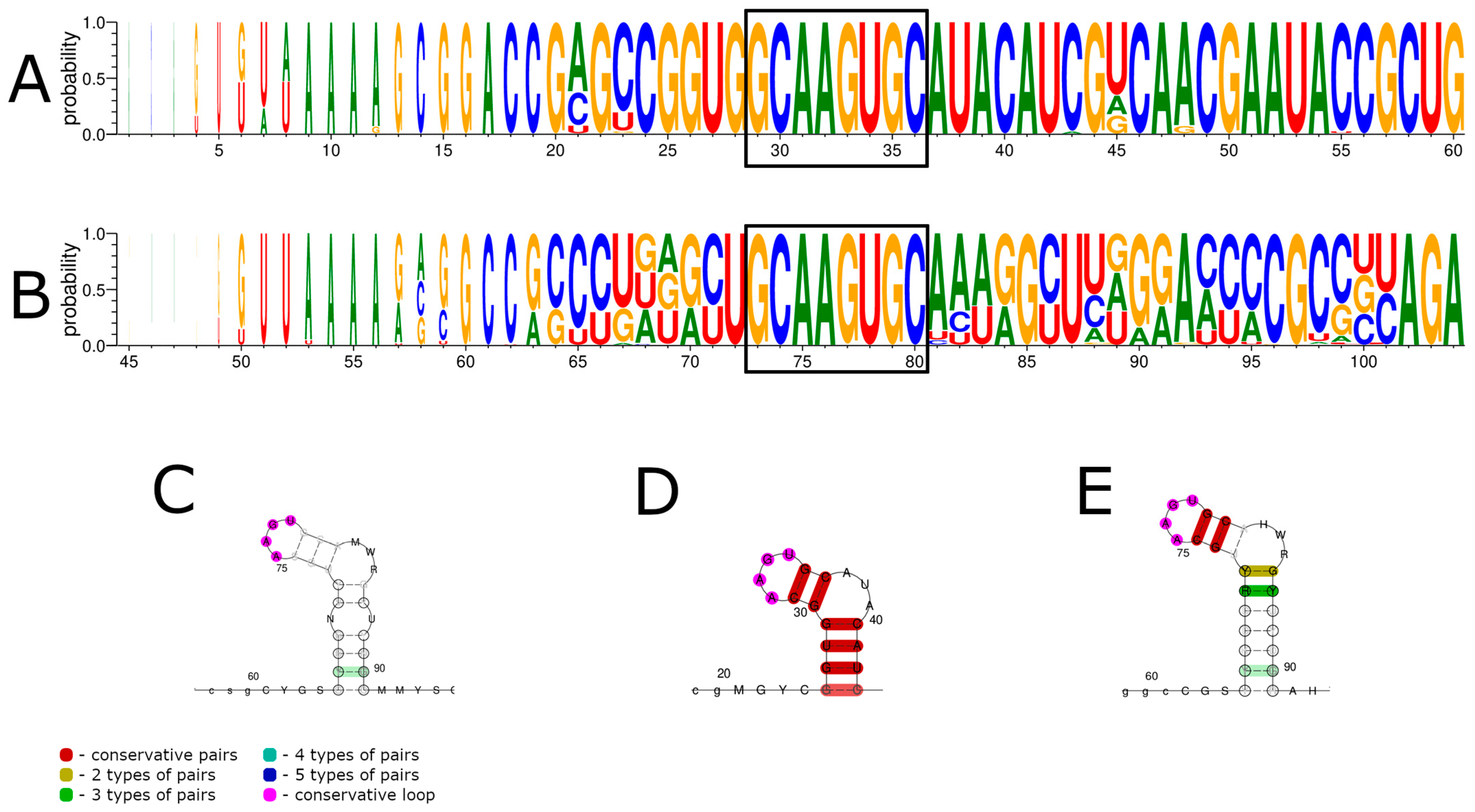
3.1. 5′ UTR Sequences and Structures
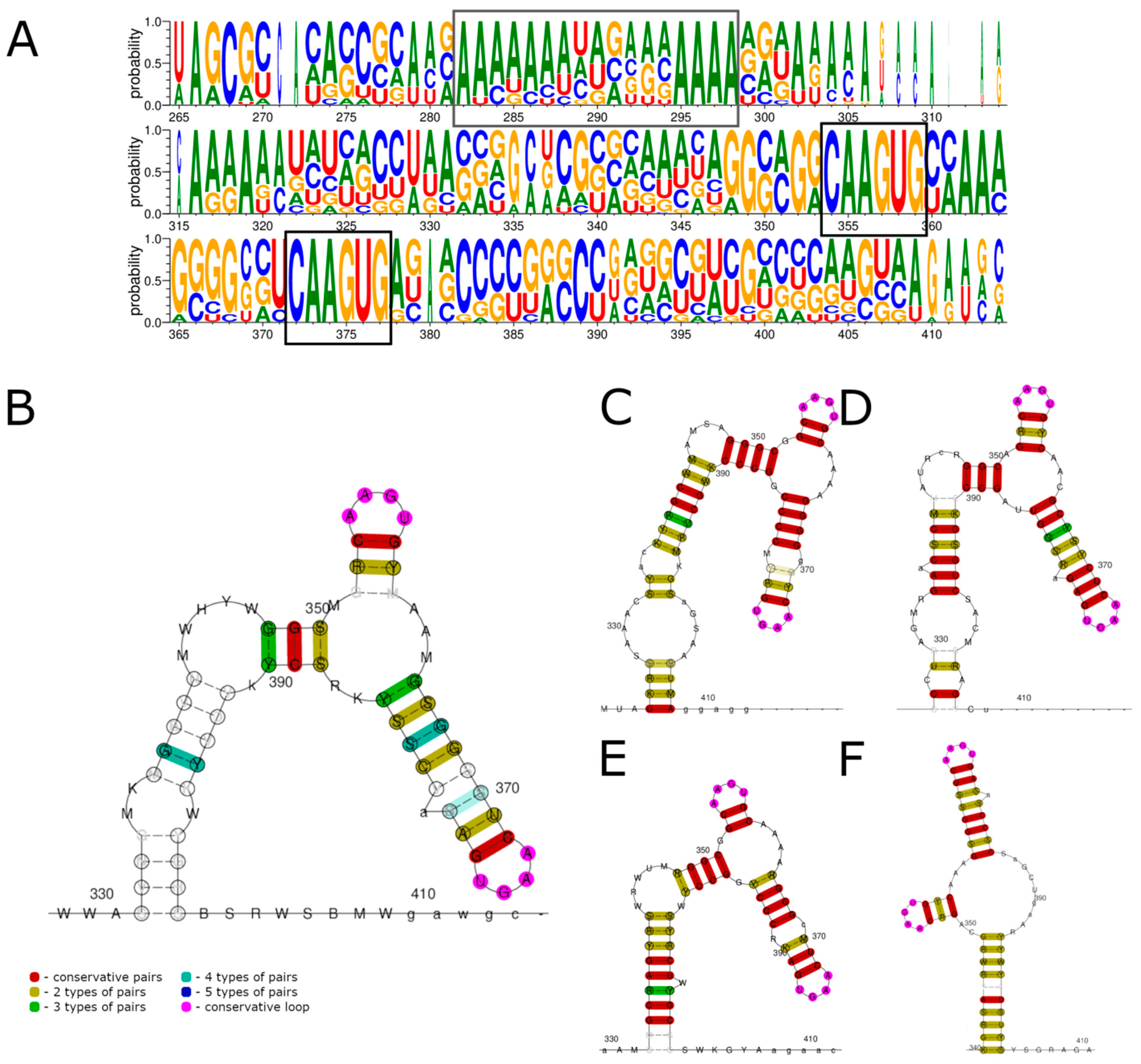
3.2. 3′ UTR Sequence and Structures
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.; Vasilakis, N.; Tian, J.; Li, C.; Chen, L.; Eastwood, G.; Diao, X.; Chen, M.-H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, M.; Lin, X.; Tian, J.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, J.; Eden, J.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Shi, M.; Tian, J.; Lin, X.; Kang, Y.; Chen, L.; Qin, X.; Xu, J.; Holmes, E.C.; Zhang, Y. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Paraskevopoulou, S.; Ka, S.; Zirkel, F.; Donath, A.; Petersen, M.; Liu, S.; Zhou, X.; Drosten, C.; Misof, B.; Junglen, S. Viromics of extant insect orders unveil the evolution of the flavi-like superfamily. Virus 2021, 7, veab030. [Google Scholar] [CrossRef]
- Kholodilov, I.S.; Litov, A.G.; Klimentov, A.S.; Belova, O.A.; Polienko, A.E.; Nikitin, N.A.; Shchetinin, A.M.; Ivannikova, A.Y.; Bell-sakyi, L.; Yakovlev, A.S.; et al. Isolation and Characterisation of Alongshan Virus in Russia. Viruses 2020, 12, 362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-D.; Wang, B.; Wei, F.; Han, S.-Z.; Zhang, L.; Yang, Z.-T.; Yan, Y.; Lv, X.-L.; Li, L.; Wang, S.-C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Kholodilov, I.S.; Belova, O.A.; Morozkin, E.S.; Litov, A.G.; Ivannikova, A.Y.; Makenov, M.T.; Shchetinin, A.M.; Aibulatov, S.V.; Bazarova, G.K.; Bell-sakyi, L.; et al. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses 2021, 13, 458. [Google Scholar] [CrossRef]
- Kobayashi, D.; Kuwata, R.; Kimura, T.; Shimoda, H.; Fujita, R.; Faizah, A.N.; Kai, I.; Matsumura, R.; Kuroda, Y.; Watanabe, S.; et al. Detection of Jingmenviruses in Japan with Evidence of Vertical Transmission in Ticks. Viruses 2021, 13, 2547. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, N.; Wang, Z.; Liu, Q. The discovery of segmented flaviviruses: Implications for viral emergence. Curr. Opin. Virol. 2020, 40, 11–18. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, J.; Zhang, L.; Zhang, Y.; Liu, L.; Niu, G. Molecular evidence for potential transovarial transmission of Jingmen tick virus in Haemaphysalis longicornis fed on cattle from Yunnan Province, China. J. Med. Virol. 2022, 95, e28357. [Google Scholar] [CrossRef]
- Dinçer, E.; Hacıoglu, S.; Kar, S.; Emanet, N.; Brinkmann, A.; Nitsche, A.; Özkul, A.; Linton, Y.-M.; Ergünay, K. Survey and Characterization of Jingmen Tick Virus Variants. Viruses 2019, 11, 1071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholodilov, I.S.; Belova, O.A.; Ivannikova, A.Y.; Gadzhikurbanov, M.N.; Makenov, M.T.; Yakovlev, A.S.; Polienko, A.E.; Dereventsova, A.V.; Litov, A.G.; Gmyl, L.V.; et al. Distribution and Characterisation of Tick-Borne Flavi-, Flavi-like, and Phenuiviruses in the Chelyabinsk Region of Russia. Viruses 2022, 14, 2699. [Google Scholar] [CrossRef]
- Villa, E.C.; Maruyama, S.R.; de Miranda-Santos, I.K.F.; Palacios, G.; Ladner, J.T. Complete Coding Genome Sequence for Mogiana Tick Virus, a Jingmenvirus Isolated from Ticks in Brazil. Genome Announc. 2017, 5, 17–18. [Google Scholar] [CrossRef] [Green Version]
- Kuivanen, S.; Levanov, L.; Kareinen, L.; Sironen, T.; Jääskeläinen, A.J.; Plyusnin, I.; Zakham, F. Detection of novel tick-borne pathogen, Alongshan virus, in Ixodes ricinus ticks, south-eastern Finland. Eurosurveillance 2019, 24, 1900394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; et al. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. mSphere 2019, 4, e00645-19. [Google Scholar] [CrossRef] [Green Version]
- Jia, N.; Liu, H.B.; Ni, X.B.; Bell-Sakyi, L.; Zheng, Y.C.; Song, J.L.; Li, J.; Jiang, B.G.; Wang, Q.; Sun, Y.; et al. Emergence of human infection with Jingmen tick virus in China: A retrospective study. eBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Souza, W.M.; Fumagalli, M.J.; Torres, A.D.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Roberto, M.; Nunes, T.; et al. Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogola, E.O.; Kopp, A.; Bastos, A.D.S.; Slothouwer, I.; Marklewitz, M.; Omoga, D.; Rotich, G.; Getugi, C.; Sang, R.; Torto, B.; et al. Jingmen Tick Virus in Ticks from Kenya. Viruses 2022, 14, 1041. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, P.; Jakupi, X.; von Possel, R.; Berisha, L.; Halili, B.; Günther, S.; Cadar, D.; Ahmeti, S.; Schmidt-Chanasit, J. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect. Genet. Evol. 2018, 65, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Domains, M.; Garry, C.E.; Garry, R.F. Proteomics Computational Analyses Suggest That the Envelope Glycoproteins of Segmented Jingmen Flavi-Like Viruses are Class II Viral Fusion Proteins. Viruses 2020, 12, 260. [Google Scholar]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-hesse, R.; Smith, D.B.; et al. ICTV ICTV Virus Taxonomy Profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Kramer, L.D.; Tesh, R.B.; Palacios, G.; Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Cell Host Microbe 2016, 20, 357–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; et al. Structures and Functions of the 3′ Untranslated Regions of Positive-Sense Single-Stranded RNA Viruses Infecting Humans and Animals. Front. Cell. Ifection Microbiol. 2020, 10, 453. [Google Scholar] [CrossRef]
- Ferron, F.; Weberc, F.; de la Torred, J.C.; Regueraa, J. Transcription and replication mechanisms of Bunyaviridae and Arenaviridae L proteins. Virus Res. 2017, 234, 118–134. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.N.; Wertz, G.W. Bunyamwera Bunyavirus RNA Synthesis Requires Cooperation of 3′- and 5′-Terminal Sequences. J. Virol. 2004, 78, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gu, M.; Zheng, Q.; Gao, R.; Liu, X. Packaging signal of influenza A virus. Virol. J. 2021, 18, 36. [Google Scholar] [CrossRef] [PubMed]
- Benavente, J.; Martinez-Costas, J. Avian reovirus: Structure and biology. Virus Res. 2007, 123, 105–119. [Google Scholar] [CrossRef]
- Chapman, E.G.; Wilusz, J.; Nix, J.C.; Kieft, J.S. The Structural Basis of Pathogenic Subgenomic Flavivirus RNA (sfRNA) Production. Science 2014, 344, 307–310. [Google Scholar] [CrossRef] [Green Version]
- Filomatori, C.V.; Lodeiro, M.F.; Alvarez, D.E.; Samsa, M.M.; Pietrasanta, L.; Gamarnik, A. V A 5′ RNA element promotes dengue virus RNA synthesis on a circular genome. GEnes Dev. 2006, 20, 2238–2249. [Google Scholar] [CrossRef] [Green Version]
- Bell-Sakyi, L. Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J. Parasitol. 1991, 77, 1006–1008. [Google Scholar] [CrossRef] [PubMed]
- Bell-Sakyi, L.; Zweygarth, E.; Blouin, E.F.; Gould, E.A.; Jongejan, F. Tick cell lines: Tools for tick and tick-borne disease research. Trends Parasitol. 2007, 23, 450–457. [Google Scholar] [CrossRef]
- Matz, M.; Shagin, D.; Bogdanova, E.; Britanova, O.; Lukyanov, S.; Diatchenko, L.; Chenchik, A. Amplification of cDNA ends based on template-switching effect and step-out PCR. Nucleic Acids Res. 1999, 27, 1558–1560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Will, S.; Joshi, T.; Hofacker, I.; Stadler, P.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner, C.; Tafer, H.; Flamm, C. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Chandonia, J. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; Mcgettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Thapar, R.; Denmon, A.P.; Nikonowicz, E.P. Recognition Modes of RNA Tetraloops And Tetraloop-Like Motifs by RNA Binding Proteins. Wiley Interdiscip. Rev. RNA 2014, 5, 49–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudin, C.; Ghazal, G.; Yoshizawa, S.; Elela, S.A.; Fourmy, D. Structure of an AAGU Tetraloop and its Contribution to Substrate Selection by yeast RNase III. J. Mol. Biol. 2006, 363, 322–331. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litov, A.G.; Okhezin, E.V.; Kholodilov, I.S.; Belova, O.A.; Karganova, G.G. Conserved Sequences in the 5′ and 3′ Untranslated Regions of Jingmenvirus Group Representatives. Viruses 2023, 15, 971. https://doi.org/10.3390/v15040971
Litov AG, Okhezin EV, Kholodilov IS, Belova OA, Karganova GG. Conserved Sequences in the 5′ and 3′ Untranslated Regions of Jingmenvirus Group Representatives. Viruses. 2023; 15(4):971. https://doi.org/10.3390/v15040971
Chicago/Turabian StyleLitov, Alexander G., Egor V. Okhezin, Ivan S. Kholodilov, Oxana A. Belova, and Galina G. Karganova. 2023. "Conserved Sequences in the 5′ and 3′ Untranslated Regions of Jingmenvirus Group Representatives" Viruses 15, no. 4: 971. https://doi.org/10.3390/v15040971