
Development and Characterization of Efficient Cell Culture Systems for Genotype 1 Hepatitis E Virus and Its Infectious cDNA Clone
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viruses
2.3. Virus Inoculation and Serial Passages
2.4. Quantification of HEV RNA
2.5. Immunocapture RT-PCR Assay
2.6. The Determination and Analysis of Full-Length and Partial Genome Sequences of JE04-1601S Strains
2.7. Construction of Full-Length Infectious cDNA Clones
2.8. In Vitro Transcription and Transfection of RNA Transcripts to PLC/PRF/5 Cells
2.9. Western Blotting
2.10. Immunofluorescence Assays
2.11. Sensitivity of HEV-1 to Ribavirin in a Cell Culture System
2.12. Lactate Dehydrogenase (LDH) Cytotoxicity Assay
2.13. Nucleotide Sequence Accession Numbers
3. Results
3.1. Serial Passages of the JE04-1601S Strain
3.2. Mutational Characteristics in Serial Passages of the JE04-1601S Strain
3.3. Replication Ability of JE04-1601S_p12 in Various Cell Lines
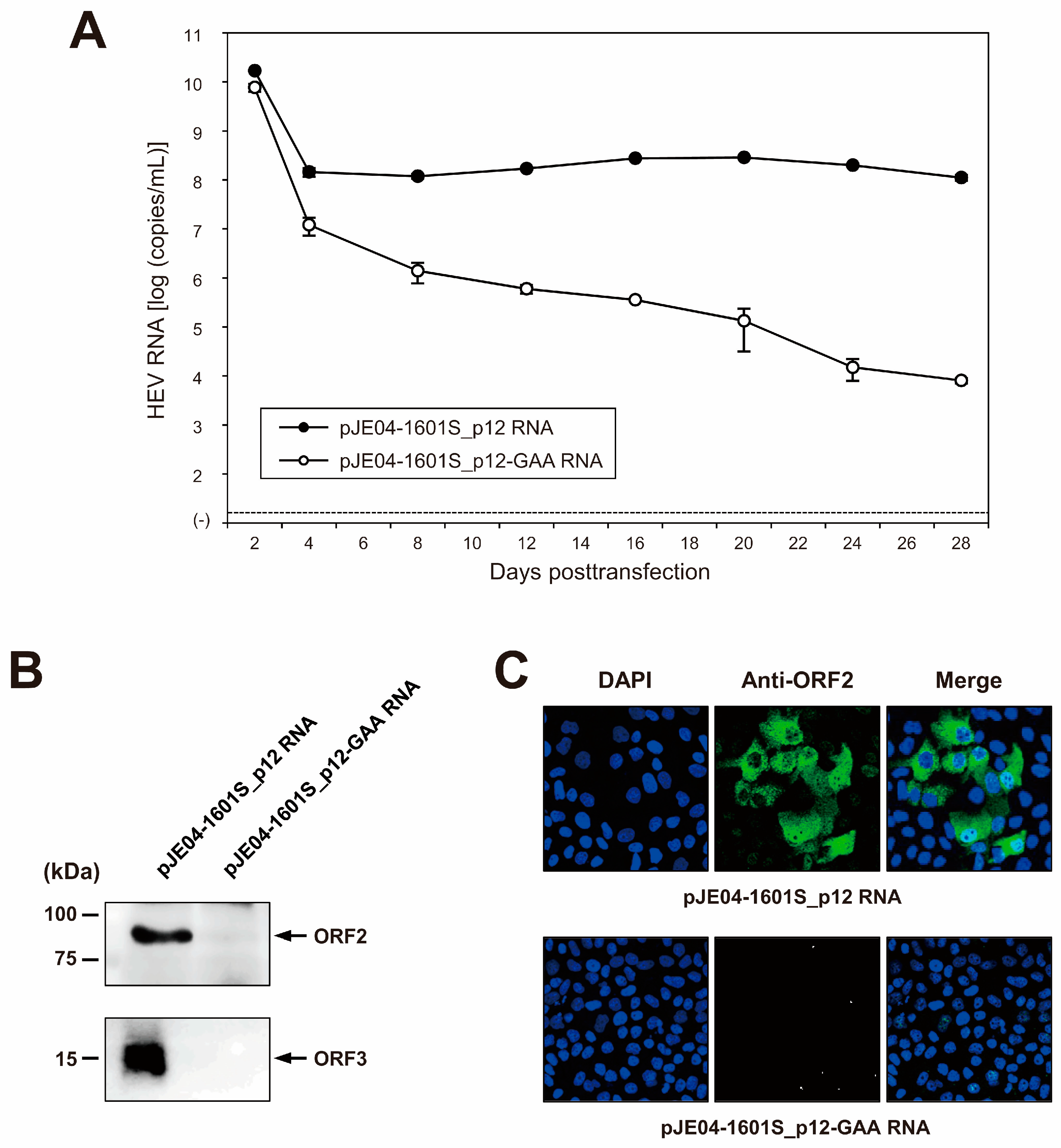
3.4. Construction of an Infectious cDNA Clone of JE04-1601S_p12 and Transfection of Its RNA Transcript to PLC/PRF/5 Cells
3.5. Characterization of cDNA-Derived JE04-1601S_p12 Progenies in the Cell Culture System
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Purdy, M.A.; Drexler, J.F.; Meng, X.J.; Norder, H.; Okamoto, H.; Van der Poel, W.H.M.; Reuter, G.; de Souza, W.M.; Ulrich, R.G.; Smith, D.B. ICTV virus taxonomy profile: Hepeviridae 2022. J. Gen. Virol. 2022, 103, 9. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Kabrane-Lazizi, Y.; Meng, X.J.; Purcell, R.H.; Emerson, S.U. Evidence that the genomic RNA of hepatitis E virus is capped. J. Virol. 1999, 73, 8848–8850. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [Green Version]
- Ichiyama, K.; Yamada, K.; Tanaka, T.; Nagashima, S.; Jirintai; Takahashi, M.; Okamoto, H. Determination of the 5’-terminal sequence of subgenomic RNA of hepatitis E virus strains in cultured cells. Arch. Virol. 2009, 154, 1945–1951. [Google Scholar] [CrossRef]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [Green Version]
- Panda, S.K.; Varma, S.P. Hepatitis E: Molecular virology and pathogenesis. J. Clin. Exp. Hepatol. 2013, 3, 114–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenney, S.P.; Meng, X.J. Hepatitis E virus genome structure and replication strategy. Cold Spring Harb. Perspect. Med. 2019, 9, a031724. [Google Scholar] [CrossRef] [PubMed]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Genetic variability and evolution of hepatitis E virus. Viruses 2019, 11, 456. [Google Scholar] [CrossRef] [Green Version]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan sulfate proteoglycans are required for cellular binding of the hepatitis E virus ORF2 capsid protein and for viral infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [Green Version]
- Xing, L.; Wang, J.C.; Li, T.C.; Yasutomi, Y.; Lara, J.; Khudyakov, Y.; Schofield, D.; Emerson, S.U.; Purcell, R.H.; Takeda, N.; et al. Spatial configuration of hepatitis E virus antigenic domain. J. Virol. 2011, 85, 1117–1124. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Nagashima, S.; Tanaka, T.; Okamoto, H. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J. Gen. Virol. 2009, 90, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Emerson, S.U.; Nguyen, H.T.; Torian, U.; Burke, D.; Engle, R.; Purcell, R.H. Release of genotype 1 hepatitis E virus from cultured hepatoma and polarized intestinal cells depends on open reading frame 3 protein and requires an intact PXXP motif. J. Virol. 2010, 84, 9059–9069. [Google Scholar] [CrossRef] [Green Version]
- Nagashima, S.; Takahashi, M.; Jirintai; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic reticulum stress induced synthesis of a novel viral factor mediates efficient replication of genotype-1 hepatitis E virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef]
- Yadav, K.K.; Boley, P.A.; Fritts, Z.; Kenney, S.P. Ectopic expression of genotype 1 hepatitis E virus ORF4 increases genotype 3 HEV viral replication in cell culture. Viruses 2021, 13, 75. [Google Scholar] [CrossRef]
- Aggarwal, R.; Goel, A. Natural history, clinical manifestations, and pathogenesis of hepatitis E virus genotype 1 and 2 infections. Cold Spring Harb. Perspect. Med. 2019, 9, a032136. [Google Scholar] [CrossRef]
- Nelson, K.E.; Labrique, A.B.; Kmush, B.L. Epidemiology of genotype 1 and 2 hepatitis E virus infections. Cold Spring Harb. Perspect. Med. 2019, 9, a031732. [Google Scholar] [CrossRef]
- Miyahara, K.; Miyake, Y.; Yasunaka, T.; Ikeda, F.; Takaki, A.; Iwasaki, Y.; Kobashi, H.; Kang, J.H.; Takahashi, K.; Arai, M.; et al. Acute hepatitis due to hepatitis E virus genotype 1 as an imported infectious disease in Japan. Intern. Med. 2010, 49, 2613–2616. [Google Scholar] [CrossRef] [Green Version]
- Nishizawa, T.; Primadharsini, P.P.; Namikawa, M.; Yamazaki, Y.; Uraki, S.; Okano, H.; Horiike, S.; Nakano, T.; Takaki, S.; Kawakami, M.; et al. Full-length genomic sequences of new subtype 1g hepatitis E virus strains obtained from four patients with imported or autochthonous acute hepatitis E in Japan. Infect. Genet. Evol. 2017, 55, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.; Mori, A.; Sugiyama, R.; Li, T.C.; Fujii, Y.; Yato, K.; Matsuda, M.; Shiota, T.; Katsumata, M.; Iwamoto, T.; et al. Isolation and genome sequencing of hepatitis E virus genotype 1 imported from India to Japan. Jpn. J. Infect. Dis. 2022, 75, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gracia, M.T.; Suay-Garcia, B.; Mateos-Lindemann, M.L. Hepatitis E and pregnancy: Current state. Rev. Med. Virol. 2017, 27, e1929. [Google Scholar] [CrossRef]
- Khuroo, M.S. Hepatitis E and pregnancy: An unholy alliance unmasked from Kashmir, India. Viruses 2021, 13, 1329. [Google Scholar] [CrossRef]
- Kamar, N.; Pischke, S. Acute and persistent hepatitis E virus genotype 3 and 4 infection: Clinical features, pathogenesis, and treatment. Cold Spring Harb. Perspect. Med. 2019, 9, a031872. [Google Scholar] [CrossRef]
- Ma, Z.; de Man, R.A.; Kamar, N.; Pan, Q. Chronic hepatitis E: Advancing research and patient care. J. Hepatol. 2022, 77, 1109–1123. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Kamar, N. Treatment of hepatitis E virus. Curr. Opin. Infect. Dis. 2016, 29, 639–644. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver. EASL clinical practice guidelines on hepatitis E virus infection. J. Hepatol. 2018, 68, 1256–1271. [Google Scholar] [CrossRef]
- Sinclair, S.M.; Jones, J.K.; Miller, R.K.; Greene, M.F.; Kwo, P.Y.; Maddrey, W.C. Final results from the ribavirin pregnancy registry, 2004–2020. Birth. Defects Res. 2022, 114, 1376–1391. [Google Scholar] [CrossRef]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antiviral. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Okamoto, H. Hepatitis E virus cell culture models. Virus Res. 2011, 161, 65–77. [Google Scholar] [CrossRef]
- Okamoto, H. Culture systems for hepatitis E virus. J. Gastroenterol. 2013, 48, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tian, D.; Sooryanarain, H.; Mahsoub, H.M.; Heffron, C.L.; Hassebroek, A.M.; Meng, X.J. Two mutations in the ORF1 of genotype 1 hepatitis E virus enhance virus replication and may associate with fulminant hepatic failure. Proc. Natl. Acad. Sci. USA 2022, 119, e2207503119. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dao Thi, V.L.; Liu, P.; Takacs, C.N.; Xiang, K.; Andrus, L.; Gouttenoire, J.; Moradpour, D.; Rice, C.M. Pan-genotype hepatitis E virus replication in stem cell-derived hepatocellular systems. Gastroenterology 2018, 154, 663–674.e7. [Google Scholar] [CrossRef] [PubMed]
- Knegendorf, L.; Drave, S.A.; Dao Thi, V.L.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef] [Green Version]
- Capelli, N.; Dubois, M.; Pucelle, M.; Da Silva, I.; Lhomme, S.; Abravanel, F.; Chapuy-Regaud, S.; Izopet, J. Optimized hepatitis E virus (HEV) culture and its application to measurements of HEV infectivity. Viruses 2020, 12, 139. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Izopet, J.; Nicot, F.; Simmonds, P.; Jameel, S.; Meng, X.J.; Norder, H.; Okamoto, H.; van der Poel, W.H.M.; Reuter, G.; et al. Update: Proposed reference sequences for subtypes of hepatitis E virus (species Orthohepevirus A). J. Gen. Virol. 2020, 101, 692–698. [Google Scholar] [CrossRef]
- Pathak, R.; Barde, P.V. Detection of genotype 1a and 1f of hepatitis E virus in patients treated at tertiary care hospitals in Central India. Intervirology 2017, 60, 201–206. [Google Scholar] [CrossRef]
- Baki, A.A.; Haque, W.; Giti, S.; Khan, A.A.; Rahman, M.M.; Jubaida, N.; Rahman, M. Hepatitis E virus genotype 1f outbreak in Bangladesh, 2018. J. Med. Virol. 2021, 93, 5177–5181. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Takahashi, M.; Kusano, E.; Okamoto, H. Development and evaluation of an efficient cell-culture system for hepatitis E virus. J. Gen. Virol. 2007, 88, 903–911. [Google Scholar] [CrossRef]
- Lorenzo, F.R.; Tanaka, T.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Yamada, K.; Inoue, J.; Takahashi, M.; Okamoto, H. Mutational events during the primary propagation and consecutive passages of hepatitis E virus strain JE03-1760F in cell culture. Virus Res. 2008, 137, 86–96. [Google Scholar] [CrossRef]
- Tanaka, T.; Takahashi, M.; Takahashi, H.; Ichiyama, K.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Okamoto, H. Development and characterization of a genotype 4 hepatitis E virus cell culture system using a HE-JF5/15F strain recovered from a fulminant hepatitis patient. J. Clin. Microbiol. 2009, 47, 1906–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Hoshino, Y.; Tanaka, T.; Takahashi, H.; Nishizawa, T.; Okamoto, H. Production of monoclonal antibodies against hepatitis E virus capsid protein and evaluation of their neutralizing activity in a cell culture system. Arch. Virol. 2008, 153, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Takahashi, M.; Nishizawa, T.; Fukai, K.; Muramatsu, U.; Yoshikawa, A. Analysis of the complete genome of indigenous swine hepatitis E virus isolated in Japan. Biochem. Biophys. Res. Commun. 2001, 289, 929–936. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Construction of an infectious cDNA clone of hepatitis E virus strain JE03-1760F that can propagate efficiently in cultured cells. J. Gen. Virol. 2009, 90, 457–462. [Google Scholar] [CrossRef]
- Sasaki, J.; Kusuhara, Y.; Maeno, Y.; Kobayashi, N.; Yamashita, T.; Sakae, K.; Takeda, N.; Taniguchi, K. Construction of an infectious cDNA clone of Aichi virus (a new member of the family Picornaviridae) and mutational analysis of a stemloop structure at the 5’ end of the genome. J. Virol. 2001, 75, 8021–8030. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Yamada, K.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch. Virol. 2008, 153, 1703–1713. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Nagashima, S.; Nishiyama, T.; Takahashi, M.; Murata, K.; Okamoto, H. Development of recombinant infectious hepatitis E virus harboring the nanoKAZ gene and its application in drug screening. J. Virol. 2022, 96, e0190621. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Nagashima, S.; Takahashi, M.; Murata, K.; Okamoto, H. Ritonavir blocks hepatitis E virus internalization and clears hepatitis E virus in vitro with ribavirin. Viruses 2022, 14, 2440. [Google Scholar] [CrossRef]
- Primadharsini, P.P.; Nagashima, S.; Okamoto, H. Mechanism of cross-species transmission, adaptive evolution and pathogenesis of hepatitis E virus. Viruses 2021, 13, 909. [Google Scholar] [CrossRef]
- Nagashima, S.; Kobayashi, T.; Tanaka, T.; Tanggis; Jirintai, S.; Takahashi, M.; Nishizawa, T.; Okamoto, H. Analysis of adaptive mutations selected during the consecutive passages of hepatitis E virus produced from an infectious cDNA clone. Virus Res. 2016, 223, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.M.; Seddik, M.I.; Gaber, M.A.; Saber, S.H.; Mandour, S.A.; El-Mokhtar, M.A. Replication of hepatitis E virus (HEV) in primary human-derived monocytes and macrophages in vitro. Vaccines 2020, 8, 239. [Google Scholar] [CrossRef] [PubMed]
- El-Mokhtar, M.A.; Othman, E.R.; Khashbah, M.Y.; Ismael, A.; Ghaliony, M.A.; Seddik, M.I.; Sayed, I.M. Evidence of the extrahepatic replication of hepatitis E virus in human endometrial stromal cells. Pathogens 2020, 9, 295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.H.; Li, X.R.; Lan, X.; Han, S.Y.; Wang, Y.N.; Hu, Y.; Pan, Q. The genetic divergences of codon usage shed new lights on transmission of hepatitis E virus from swine to human. Infect. Genet. Evol. 2019, 68, 23–29. [Google Scholar] [CrossRef]
- Sun, J.; Ren, C.; Huang, Y.; Chao, W.; Xie, F. The effects of synonymous codon usages on genotypic formation of open reading frames in hepatitis E virus. Infect. Genet. Evol. 2020, 85, 104450. [Google Scholar] [CrossRef]
- Feagins, A.R.; Cordoba, L.; Sanford, B.J.; Dryman, B.A.; Huang, Y.W.; LeRoith, T.; Emerson, S.U.; Meng, X.J. Intergenotypic chimeric hepatitis E viruses (HEVs) with the genotype 4 human HEV capsid gene in the backbone of genotype 3 swine HEV are infectious in pigs. Virus Res. 2011, 156, 141–146. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, S.N.; Devhare, P.B.; Pingle, S.Y.; Paingankar, M.S.; Arankalle, V.A.; Lole, K.S. Hepatitis E virus (HEV)-1 harbouring HEV-4 non-structural protein (ORF1) replicates in transfected porcine kidney cells. J. Gen. Virol. 2016, 97, 1829–1840. [Google Scholar] [CrossRef] [Green Version]
- Cordoba, L.; Feagins, A.R.; Opriessnig, T.; Cossaboom, C.M.; Dryman, B.A.; Huang, Y.W.; Meng, X.J. Rescue of a genotype 4 human hepatitis E virus from cloned cDNA and characterization of intergenotypic chimeric viruses in cultured human liver cells and in pigs. J. Gen. Virol. 2012, 93, 2183–2194. [Google Scholar] [CrossRef] [Green Version]
- Tian, D.; Yugo, D.M.; Kenney, S.P.; Lynn Heffron, C.; Opriessnig, T.; Karuppannan, A.K.; Bayne, J.; Halbur, P.G.; Meng, X.J. Dissecting the potential role of hepatitis E virus ORF1 nonstructural gene in cross-species infection by using intergenotypic chimeric viruses. J. Med. Virol. 2020, 92, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223.e8. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Passage | Inoculum | Cells | Viral Load of HEV Inoculated in Each Well (Copies Per Well) a |
|---|---|---|---|
| 0 | Serum (JE04-1601S_wt) | PLC/PRF/5 | 1.5 × 106 |
| 1 | Culture supernatant (26th day after the first inoculation) | A549_1-1H8 | 1.1 × 104 |
| 2 | Culture supernatant (62nd day after the second inoculation) | A549_1-1H8 | 4.8 × 104 |
| 3 | Culture supernatant (42nd day after the third inoculation) | A549_1-1H8 | 1.0 × 105 |
| 4 | Culture supernatant (42nd day after the fourth inoculation) | A549_1-1H8 | 6.8 × 104 |
| 5 | Culture supernatant (70th day after the fifth inoculation) | A549_1-1H8 | 1.0 × 105 |
| 6 | Culture supernatant (54th day after the sixth inoculation) | A549_1-1H8 | 1.0 × 105 |
| 7 | Culture supernatant (64th day after the seventh inoculation) | A549_1-1H8 | 1.0 × 105 |
| 8 | Culture supernatant (30th day after the eighth inoculation) | A549_1-1H8 | 1.0 × 105 |
| 9 | Culture supernatant (36th day after the ninth inoculation) | A549_1-1H8 | 1.0 × 105 |
| 10 | Culture supernatant (26th day after the tenth inoculation) | A549_1-1H8 | 1.0 × 105 |
| 11 | Culture supernatant (54th day after the eleventh inoculation) | A549_1-1H8 | 1.0 × 105 |
| 12 | Culture supernatant (38th day after the twelfth inoculation) | A549_1-1H8 | 1.0 × 105 |
| Name | Polarity | Sequence (5′ to 3′) | Note |
|---|---|---|---|
| SSP-T | AAGGATCCGTCGACATCGATAATACGTTTTTTTTTTTTTTT | cDNA synthesis | |
| 1601S-1 | + | GCTTAATACGACTCACTATAGCAGACCACATATGTGGTCGATGCC | T7 promoter (underlined) and positive-strand sequence (nt 1–25 a) |
| 1601S-2 | – | TTGCATCGGAGATGCCCACCTCG | Negative-strand sequence (nt 3598–3620) |
| 1601S-3 | + | GGTGGGCATCTCCGATGCAATCG | Positive-strand sequence (nt 3601–3623) |
| 1601S-4 | – | GCCCCAAGGGGTTATGCTAGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTCAGGGAGCGCGAAACGCAGAAAAGAG | pUC19 vector, poly(A), and negative-strand sequence (nt 7167–7192) |
| 1601S-5 | + | CTAGCATAACCCCTTGGGGCCTC | pUC19 vector |
| 1601S-6 | – | TATAGTGAGTCGTATTAAGCTTGGCG | T7 promoter (underlined) and pUC19 vector |
| 1601S-7 | – | ACAGAATGGATTGGCCGACTCCC | Negative-strand sequence (nt 2088–2110) |
| 1601S-8 | + | AGTCGGCCAATCCATTCTGTGGC | Positive-strand sequence (nt 2091–2113) |
| 1601S-9 | + | GGTGGGCATCTCCGATGCAACTCTAAACGGGTCTTGAGGGG | Positive-strand sequence (nt 3601–3620) and pUC19 vector |
| 1601S-SpeI-F | + | TTGGGCAGAAACTAGTGTTCACCC | Positive-strand sequence (nt 3384–3407), SpeI site (underlined) |
| 1601S-GAA-R | – | ATCGAGGCGGCACCTTTGAAGGCAGCCACCTGC | Negative-strand sequence (nt 4654–4686), mutated nucleotide (underlined) |
| 1601S-GAA-F | + | AGGTGCCGCCTCGATAGTGCTTTGCAGTGAGTACC | Positive-strand sequence (nt 4672–4706), mutated nucleotide (underlined) |
| 1601S-SpeI-R | – | CGCCATTAGTACTAGTAAAATAAAGATCC | Negative-strand sequence (nt 6197–6225), SpeI site (underlined) |
| Nucleotide Position | Region (Domain) a | Nucleotide b | Amino Acid | |||
|---|---|---|---|---|---|---|
| JE04-1601S_wt | Passage 10 (p10) | Passage 12 (p12) | Position | Substitution | ||
| 244 | ORF1 (MeT) | C | T | T | 73 | - |
| 433 | ORF1 (MeT) | C | C | T | 136 | - |
| 477 | ORF1 (MeT) | C | C | T | 151 | Ser to Phe |
| 617 | ORF1 (MeT) | C | T | T | 198 | - |
| 895 | ORF1 (Y) | A | A | G | 290 | - |
| 1270 | ORF1 (Y) | C | T | T | 415 | - |
| 1738 | ORF1 (PCP) | C | T | T | 571 | - |
| 1870 | ORF1 (PCP) | T | C | C | 615 | - |
| 2311 | ORF1 (HVR) | C | T | T | 762 | - |
| 2356 | ORF1 (X) | T | C | C | 777 | - |
| 2728 | ORF1 (X) | C | T | T | 901 | - |
| 2988 | ORF1 (Hel) | C | G | G | 988 | Ala to Gly |
| 3112 | ORF1 (Hel) | T | C | C | 1029 | - |
| 3298 | ORF1 (Hel) | T | T | C | 1091 | - |
| 3667 | ORF1 (RdRp) | C | Yb | C | 1214 | - |
| 4417 | ORF1 (RdRp) | T | C | C | 1464 | - |
| 4603 | ORF1 (RdRp) | T | T | C | 1526 | - |
| 5090 | ORF1 (RdRp) | C | Y | T | 1689 | - |
| 5528 | ORF2 | C | Y | T | 128 | - |
| 6613 | ORF2 | G | G | C | 490 | Gly to Ala |
| 6770 | ORF2 | C | C | T | 542 | - |
| Wild-Type or Variants of JE04-1601S | Nucleotide Position | ||
|---|---|---|---|
| 2728 | 2988 | 3112 | |
| Wild-type (serum) | C | C | T |
| p0 (culture supernatant, 26 dpi a) | C | C | C |
| p1 (culture supernatant, 62 dpi) | T | C | C |
| p2 (culture supernatant, 42 dpi) | T | C | C |
| p3 (culture supernatant, 60 dpi) | T | C | C |
| p4 (culture supernatant, 70 dpi) | T | S b | C |
| p5 (culture supernatant, 54 dpi) | T | G | C |
| p6 (culture supernatant, 64 dpi) | T | G | C |
| p7 (culture supernatant, 30 dpi) | T | G | C |
| p8 (culture supernatant, 36 dpi) | T | G | C |
| p9 (culture supernatant, 26 dpi) | T | G | C |
| p10 (culture supernatant, 54 dpi) | T | G | C |
| p11 (culture supernatant, 38 dpi) | T | G | C |
| p12 (culture supernatant, 26 dpi) | T | G | C |
| Inoculated Cells | % of Captured HEV Particles a in the Total HEV-1 Per Tube | |
|---|---|---|
| 20 Days Postinoculation | 60 Days Postinoculation | |
| PLC/PRF/5 | 98.7 | 95.8 |
| A549_1-1H8 | 98.9 | 98.5 |
| HepG2/C3A | 95.6 | 100.0 |
| PK15 | 10.1 | N/A b |
| IBRS-2 | 12.3 | N/A |
| LLC-PK1 | 20.5 | N/A |
| Treatment | LDH Release (Mean ± SD) a | |
|---|---|---|
| 12 Days Postinoculation | 28 Days Postinoculation | |
| No drug treatment | 2.4% ± 0.3% | 2.4% ± 0.3% |
| 40 μM Ribavirin | 2.1% ± 0.4% | 2.4% ± 0.3% |
| 160 μM Ribavirin | 2.8% ± 0.3% | 3.7% ± 0.3% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primadharsini, P.P.; Nagashima, S.; Tanaka, T.; Jirintai, S.; Takahashi, M.; Murata, K.; Okamoto, H. Development and Characterization of Efficient Cell Culture Systems for Genotype 1 Hepatitis E Virus and Its Infectious cDNA Clone. Viruses 2023, 15, 845. https://doi.org/10.3390/v15040845
Primadharsini PP, Nagashima S, Tanaka T, Jirintai S, Takahashi M, Murata K, Okamoto H. Development and Characterization of Efficient Cell Culture Systems for Genotype 1 Hepatitis E Virus and Its Infectious cDNA Clone. Viruses. 2023; 15(4):845. https://doi.org/10.3390/v15040845
Chicago/Turabian StylePrimadharsini, Putu Prathiwi, Shigeo Nagashima, Toshinori Tanaka, Suljid Jirintai, Masaharu Takahashi, Kazumoto Murata, and Hiroaki Okamoto. 2023. "Development and Characterization of Efficient Cell Culture Systems for Genotype 1 Hepatitis E Virus and Its Infectious cDNA Clone" Viruses 15, no. 4: 845. https://doi.org/10.3390/v15040845