
Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors
 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Identification of Pan-Coronavirus Inhibitors
2.2. Antiviral Activity and Cytotoxicity of the NBCoV Small Molecules in a Pseudovirus Assay
2.3. Antiviral Activity of NBCoV35-37 and NBCoV63 against SARS-CoV-2 Mutant (D614G) and Four VOCs
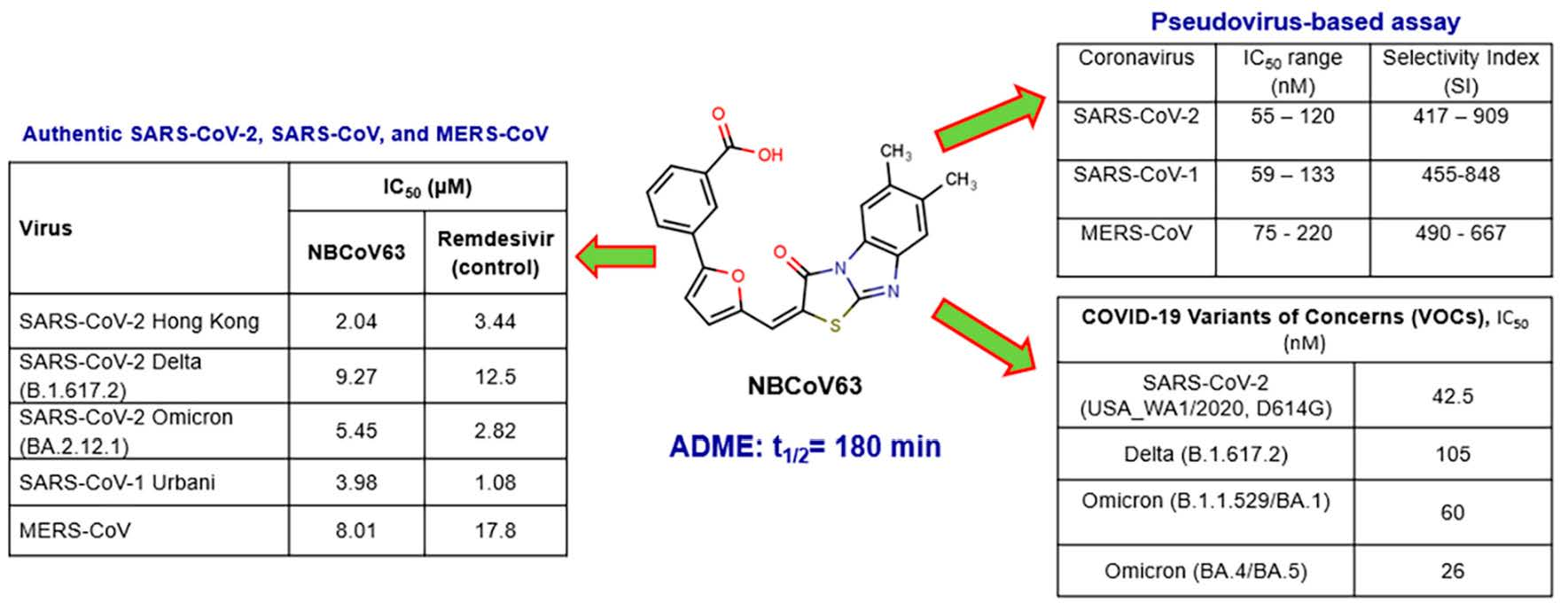
2.4. NBCoV63 Inhibited Three Variants of the Replication-Competent Authentic Virus SARS-CoV-2 and Two Closely Related Highly Pathogenic Coronaviruses, SARS-CoV-1 and MERS-CoV
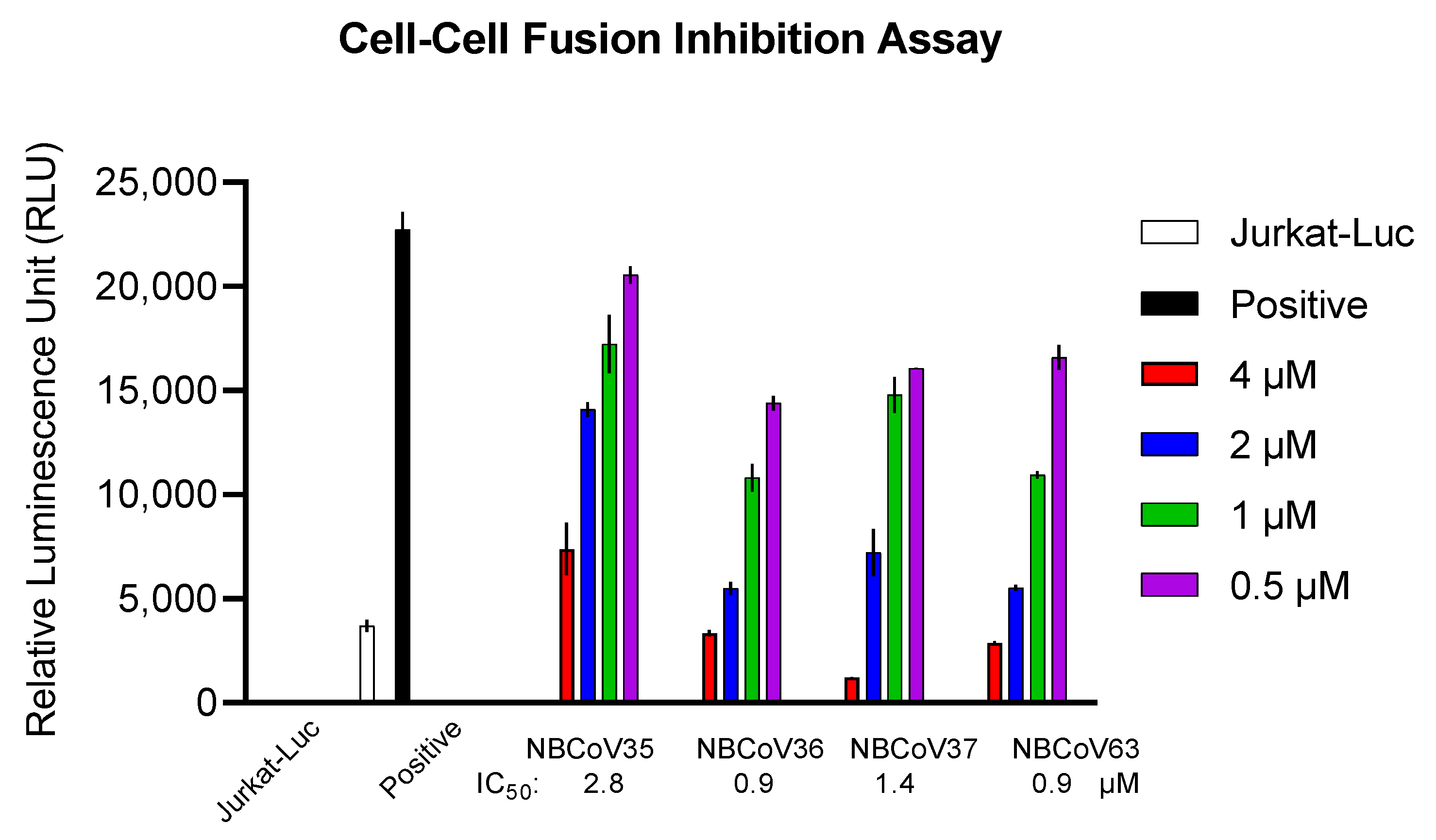
2.5. NBCoV Small Molecules Inhibited the SARS-CoV-2 Mediated Cell-to-Cell Fusion
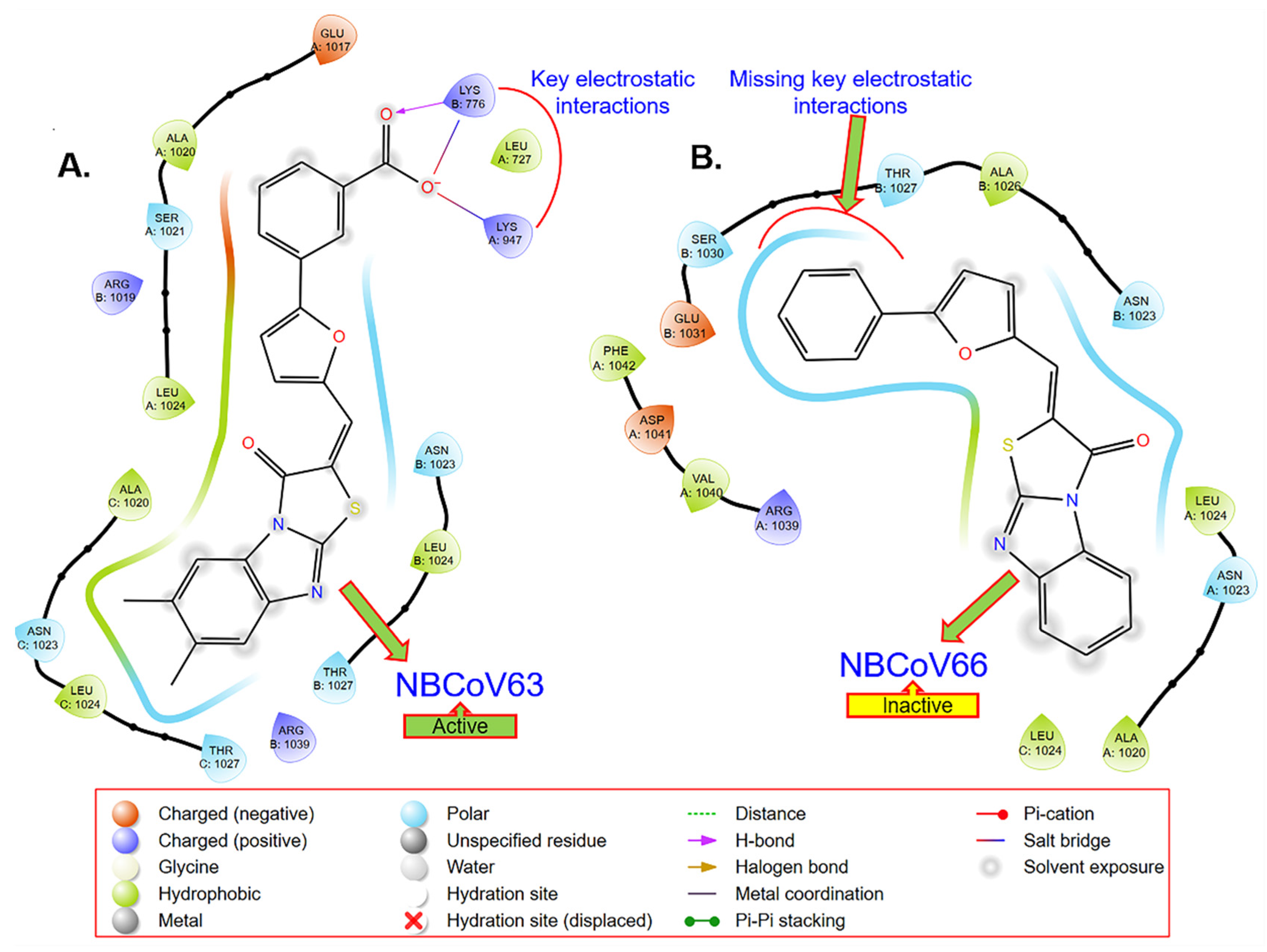
2.6. Computer-Based Determination of the Binding Site of NBCoV63 in the SARS-CoV-2 Spike Protein
2.7. In Vitro ADME
3. Conclusions
4. Experimental Section
4.1. Cells and Plasmids
4.2. Small Molecules
4.3. Pseudoviruses Preparation
4.4. Measurement of Antiviral Activity
4.5. Evaluation of Cytotoxicity
4.6. Drug Sensitivity of Spike-Mutated Pseudovirus
4.7. Cell-to-Cell Fusion Inhibition Assay
4.8. GLIDE-Based Docking
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Phelan, A.L.; Katz, R.; Gostin, L.O. The Novel Coronavirus Originating in Wuhan, China: Challenges for Global Health Governance. JAMA 2020, 323, 709–710. [Google Scholar] [CrossRef] [PubMed]
- Butt, A.A.; Khan, T.; Yan, P.; Shaikh, O.S.; Omer, S.B.; Mayr, F. Rate and risk factors for breakthrough SARS-CoV-2 infection after vaccination. J. Infect. 2021, 83, 237–279. [Google Scholar] [CrossRef]
- Shastri, J.; Parikh, S.; Aggarwal, V.; Agrawal, S.; Chatterjee, N.; Shah, R.; Devi, P.; Mehta, P.; Pandey, R. Severe SARS-CoV-2 Breakthrough Reinfection with Delta Variant after Recovery from Breakthrough Infection by Alpha Variant in a Fully Vaccinated Health Worker. Front. Med. 2021, 8, 737007. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Wyka, K.; White, T.M.; Picchio, C.A.; Rabin, K.; Ratzan, S.C.; Parsons Leigh, J.; Hu, J.; El-Mohandes, A. Revisiting COVID-19 vaccine hesitancy around the world using data from 23 countries in 2021. Nat. Commun. 2022, 13, 3801. [Google Scholar] [CrossRef]
- Gandhi, S.; Klein, J.; Robertson, A.J.; Peña-Hernández, M.A.; Lin, M.J.; Roychoudhury, P.; Lu, P.; Fournier, J.; Ferguson, D.; Mohamed Bakhash, S.A.K.; et al. De novo emergence of a remdesivir resistance mutation during treatment of persistent SARS-CoV-2 infection in an immunocompromised patient: A case report. Nat. Commun. 2022, 13, 1547. [Google Scholar] [CrossRef]
- Hogan, J.I.; Duerr, R.; Dimartino, D.; Marier, C.; Hochman, S.E.; Mehta, S.; Wang, G.; Heguy, A. Remdesivir Resistance in Transplant Recipients With Persistent Coronavirus Disease 2019 (COVID-19). Clin. Infect. Dis. 2023, 76, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Service, R.F. Bad news for Paxlovid? Resistance may be coming. Science 2022, 377, 138–139. [Google Scholar] [CrossRef]
- Chen, R.E.; Zhang, X.; Case, J.B.; Winkler, E.S.; Liu, Y.; VanBlargan, L.A.; Liu, J.; Errico, J.M.; Xie, X.; Suryadevara, N.; et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat. Med. 2021, 27, 717–726. [Google Scholar] [CrossRef]
- Diamond, M.; Chen, R.; Xie, X.; Case, J.; Zhang, X.; VanBlargan, L.; Liu, Y.; Liu, J.; Errico, J.; Winkler, E.; et al. SARS-CoV-2 variants show resistance to neutralization by many monoclonal and serum-derived polyclonal antibodies. Res. Sq. 2021, 1, 1–17. [Google Scholar]
- Focosi, D.; Maggi, F. Neutralising antibody escape of SARS-CoV-2 spike protein: Risk assessment for antibody-based COVID-19 therapeutics and vaccines. Rev. Med. Virol. 2021, 31, e2231. [Google Scholar] [CrossRef]
- Weisblum, Y.; Schmidt, F.; Zhang, F.; DaSilva, J.; Poston, D.; Lorenzi, J.C.; Muecksch, F.; Rutkowska, M.; Hoffmann, H.H.; Michailidis, E.; et al. Escape from neutralizing antibodies by SARS-CoV-2 spike protein variants. eLife 2020, 9, e61312. [Google Scholar] [CrossRef]
- Mahase, E. Delta variant: What is happening with transmission, hospital admissions, and restrictions? BMJ 2021, 373, n1513. [Google Scholar] [CrossRef]
- Reardon, S. How the Delta variant achieves its ultrafast spread. Nature 2021, 21, 1–3. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2022, 186, 279–286. [Google Scholar] [CrossRef]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Zamorano Cuervo, N.; Grandvaux, N. ACE2: Evidence of role as entry receptor for SARS-CoV-2 and implications in comorbidities. eLife 2020, 9, e61390. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Muller, M.A.; Allen, P.; Soilleux, E.; Pfefferle, S.; Steffen, I.; Tsegaye, T.S.; He, Y.; Gnirss, K.; et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011, 85, 4122–4134. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Nagata, N.; Shirato, K.; Kawase, M.; Takeda, M.; Taguchi, F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010, 84, 12658–12664. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Lu, G.; Qi, J.; Li, Y.; Wu, Y.; Deng, Y.; Geng, H.; Li, H.; Wang, Q.; Xiao, H.; et al. Structure of the fusion core and inhibition of fusion by a heptad repeat peptide derived from the S protein of Middle East respiratory syndrome coronavirus. J. Virol. 2013, 87, 13134–13140. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Xia, X. Domains and functions of spike protein in SARS-Cov-2 in the context of vaccine design. Viruses 2021, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Gangadevi, S.; Badavath, V.N.; Thakur, A.; Yin, N.; De Jonghe, S.; Acevedo, O.; Jochmans, D.; Leyssen, P.; Wang, K.; Neyts, J.; et al. Kobophenol A inhibits binding of host ACE2 receptor with spike RBD domain of SARS-CoV-2, a lead compound for blocking COVID-19. J. Phys. Chem. Lett. 2021, 12, 1793–1802. [Google Scholar] [CrossRef]
- Larue, R.C.; Xing, E.; Kenney, A.D.; Zhang, Y.; Tuazon, J.A.; Li, J.; Yount, J.S.; Li, P.K.; Sharma, A. Rationally designed ACE2-derived peptides inhibit SARS-CoV-2. Bioconjug. Chem. 2021, 32, 215–223. [Google Scholar] [CrossRef]
- Linsky, T.W.; Vergara, R.; Codina, N.; Nelson, J.W.; Walker, M.J.; Su, W.; Barnes, C.O.; Hsiang, T.Y.; Esser-Nobis, K.; Yu, K.; et al. De novo design of potent and resilient hACE2 decoys to neutralize SARS-CoV-2. Science 2020, 370, 1208–1214. [Google Scholar] [CrossRef]
- Sitthiyotha, T.; Chunsrivirot, S. Computational design of 25-mer peptide binders of SARS-CoV-2. J. Phys. Chem. B 2020, 124, 10930–10942. [Google Scholar] [CrossRef]
- Yang, J.; Petitjean, S.J.L.; Koehler, M.; Zhang, Q.; Dumitru, A.C.; Chen, W.; Derclaye, S.; Vincent, S.P.; Soumillion, P.; Alsteens, D. Molecular interaction and inhibition of SARS-CoV-2 binding to the ACE2 receptor. Nat. Commun. 2020, 11, 4541. [Google Scholar] [CrossRef]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell. Mol. Immunol. 2020, 17, 765–767. [Google Scholar] [CrossRef]
- Curreli, F.; Victor, S.M.B.; Ahmed, S.; Drelich, A.; Tong, X.; Tseng, C.K.; Hillyer, C.D.; Debnath, A.K. Stapled peptides based on human angiotensin-converting enzyme 2 (ACE2) potently inhibit SARS-CoV-2 infection in vitro. mBio 2020, 11, e02451-20. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Carter, L.; Walls, A.C.; Park, Y.J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef] [PubMed]
- Ou, X.; Liu, Y.; Lei, X.; Li, P.; Mi, D.; Ren, L.; Guo, L.; Guo, R.; Chen, T.; Hu, J.; et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020, 11, 1620. [Google Scholar] [CrossRef] [PubMed]
- Klasse, P.J.; Moore, J.P. Antibodies to SARS-CoV-2 and their potential for therapeutic passive immunization. eLife 2020, 9, e57877. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Mutations in the SARS-CoV-2 spike RBD are responsible for stronger ACE2 binding and poor anti-SARS-CoV mAbs cross-neutralization. Comput. Struct. Biotechnol. J. 2020, 18, 3402–3414. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294.e9. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; van der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef]
- Williams, T.C.; Burgers, W.A. SARS-CoV-2 evolution and vaccines: Cause for concern? Lancet Respir. Med. 2021, 9, 333–335. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Addetia, A.; Hannon, W.W.; Choudhary, M.C.; Dingens, A.S.; Li, J.Z.; Bloom, J.D. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021, 371, 850–854. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Liu, Q.; Zhu, Y.; Chan, K.H.; Qin, L.; Li, Y.; Wang, Q.; Chan, J.F.; Du, L.; Yu, F.; et al. Structure-based discovery of Middle East respiratory syndrome coronavirus fusion inhibitor. Nat. Commun. 2014, 5, 3067. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Liu, S.; Li, J.; Lu, H.; Qi, Z.; Liu, Z.; Debnath, A.K.; Jiang, S. Conserved salt bridge between the N- and C-terminal heptad repeat regions of the human immunodeficiency virus type 1 gp41 core structure is critical for virus entry and inhibition. J. Virol. 2008, 82, 11129–11139. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.Y.; Sivapackiam, J.; Pandya, K.; Jiang, S.; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar] [CrossRef] [PubMed]
- Curreli, F.; Ahmed, S.; Victor, S.M.B.; Drelich, A.; Panda, S.S.; Altieri, A.; Kurkin, A.V.; Tseng, C.K.; Hillyer, C.D.; Debnath, A.K. Discovery of Highly Potent Fusion Inhibitors with Potential Pan-Coronavirus Activity That Effectively Inhibit Major COVID-19 Variants of Concern (VOCs) in Pseudovirus-Based Assays. Viruses 2021, 14, 69. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, I.; Vilhardt, F. Macropinocytosis is the entry mechanism of amphotropic murine leukemia virus. J. Virol. 2015, 89, 1851–1866. [Google Scholar] [CrossRef]
- Mothes, W.; Sherer, N.M.; Jin, J.; Zhong, P. Virus cell-to-cell transmission. J. Virol. 2010, 84, 8360–8368. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Debnath, A.K.; Radigan, L.; Jiang, S. Structure-based identification of small molecule antiviral compounds targeted to the gp41 core structure of the human immunodeficiency virus type 1. J. Med. Chem. 1999, 42, 3203–3209. [Google Scholar] [CrossRef]
- Ji, H.; Shu, W.; Burling, T.; Jiang, S.; Lu, M. Inhibition of HIV-1 infectivity by the gp41 core: Role of a conserved hydrophobic cavity in membrane fusion. J. Virol. 1999, 73, 8578–8586. [Google Scholar] [CrossRef]
- Jiang, S.; Debanth, A.K. The HIV-1 gp41 core: A target for developing HIV-1 inhibitors. Curr. Top. Biochem. Res. 2000, 2, 1–17. [Google Scholar]
- Jiang, S.; Debnath, A.K. Development of HIV entry inhibitors targeted to the coiled coil regions of gp41. Biochem. Biophys. Res. Commun. 2000, 269, 641–646. [Google Scholar] [CrossRef] [PubMed]
- Ernst, J.T.; Kutzki, O.; Debnath, A.K.; Jiang, S.; Lu, H.; Hamilton, A.D. Design of a protein surface antagonist based on alpha-helix mimicry: Inhibition of gp41 assembly and viral fusion. Angew. Chem. Int. Ed. Engl. 2002, 41, 278–281. [Google Scholar] [CrossRef]
- Waring, M.J.; Arrowsmith, J.; Leach, A.R.; Leeson, P.D.; Mandrell, S.; Owen, R.M.; Pairaudeau, G.; Pennie, W.D.; Pickett, S.D.; Wang, J.; et al. An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat. Rev. Drug Discov. 2015, 14, 475–486. [Google Scholar] [CrossRef]
- Smith, D.A.; Di, L.; Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: Misconceptions in drug discovery. Nat. Rev. Drug Discov. 2010, 9, 929–939. [Google Scholar] [CrossRef]
- Backman, J.T.; Filppula, A.M.; Niemi, M.; Neuvonen, P.J. Role of Cytochrome P450 2C8 in Drug Metabolism and Interactions. Pharmacol. Rev. 2016, 68, 168–241. [Google Scholar] [CrossRef]
- Turpeinen, M.; Zanger, U.M. Cytochrome P450 2B6: Function, genetics, and clinical relevance. Drug Metabol. Drug Interact. 2012, 27, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 1997, 2, 436–444. [Google Scholar] [CrossRef]
- Schmidt, F.; Weisblum, Y.; Muecksch, F.; Hoffmann, H.H.; Michailidis, E.; Lorenzi, J.C.C.; Mendoza, P.; Rutkowska, M.; Bednarski, E.; Gaebler, C.; et al. Measuring SARS-CoV-2 neutralizing antibody activity using pseudotyped and chimeric viruses. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Chang, L.J.; Urlacher, V.; Iwakuma, T.; Cui, Y.; Zucali, J. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther. 1999, 6, 715–728. [Google Scholar] [CrossRef] [PubMed]
- Landau, N.R.; Page, K.A.; Littman, D.R. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency virus host range. J. Virol. 1991, 65, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef]
- He, J.; Choe, S.; Walker, R.; di Marzio, P.; Morgan, D.O.; Landau, N.R. Human immunodeficiency virus type 1 viral protein R (Vpr) arrests cells in the G2 phase of the cell cycle by inhibiting p34cdc2 activity. J. Virol. 1995, 69, 6705–6711. [Google Scholar] [CrossRef] [PubMed]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche. Naunyn Schmiedebergs Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Nie, J.; Li, Q.; Wu, J.; Zhao, C.; Hao, H.; Liu, H.; Zhang, L.; Nie, L.; Qin, H.; Wang, M.; et al. Establishment and validation of a pseudovirus neutralization assay for SARS-CoV-2. Emerg. Microbes Infect. 2020, 9, 680–686. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Compounds | Structure | 293T/ACE2 Cells | A549/ACE2/TMPRSS2 Cells | ||||
|---|---|---|---|---|---|---|---|
| IC50 (nM) a | CC50 (µM) a | SI | IC50 (nM) a | CC50 (µM) a | SI | ||
| NBCoV35 |  | 198 ± 9 | ~50 | 253 | 126 ± 3 | ~50 | 397 |
| NBCoV36 |  | 330 ± 18 | 43 ± 0.9 | 130 | 227 ± 18 | >50 | >220 |
| NBCoV37 |  | 66 ± 2 | 42.5 ± 2 | 644 | 145 ± 12 | ~50 | 345 |
| NBCoV62 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV63 |  | 80 ± 7 | >50 | >625 | 55 ± 3 | ~50 | 909 |
| NBCoV65 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV66 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV68 |  | 3900 ± 100 | >50 | >13 | 2300 ± 70 | >50 | >22 |
| NBCoV69 |  | 2550 ± 105 | >50 | >20 | 3000 ± 400 | >50 | >17 |
| NBCoV70 |  | 9500 ± 1000 | >50 | >5 | >10,000 | >50 | ND |
| NBCoV71 |  | >10,000 | 48 ± 2 | ND | >10,000 | 40 ± 2 | ND |
| NBCoV72 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV73 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV74 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV75 |  | 5200 ± 600 | >50 | >10 | 3300 ± 700 | >50 | >15 |
| NBCoV76 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV77 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV78 |  | >10,000 | >50 | ND | 8100 ± 200 | >50 | >6 |
| NBCoV79 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV80 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| NBCoV81 |  | 387 ± 64 | >50 | >129 | 459 ± 23 | >50 | >109 |
| NBCoV82 |  | >10,000 | >50 | ND | >10,000 | >50 | ND |
| Compounds | SARS-CoV-2 IC50 (nM) a | ||||
|---|---|---|---|---|---|
| USA-WA1/2020 D614G | Variants of Concern (VOC) | ||||
| Delta (B.1.617.2) | Omicron (B.1.1.529/BA.1) | Omicron (BA.4/BA.5) | Brazil (Gamma: K417T/E484K/N501Y) | ||
| 293T/ACE2 Cells | |||||
| NBCoV35 | 195 ± 10 | 335 ± 5 | 173 ± 30 | 94 ± 6 | 506 ± 44 |
| NBCoV36 | 430 ± 31 | 802 ± 143 | 368 ± 90 | 181 ± 12 | 1183 ± 58 |
| NBCoV37 | 157 ± 1.5 | 267 ± 23 | 135 ± 3 | 82 ± 8 | 260 ± 52 |
| NBCoV63 | 40.8 ± 1.2 | 84 ± 0.5 | 96 ± 11 | 34 ± 1 | 77.5 ± 17.3 |
| A549/ACE2/TMPRSS2 Cells | |||||
| NBCoV35 | 266 ± 15 | 236 ± 6 | 201 ± 38 | 170 ± 13 | 292 ± 23 |
| NBCoV36 | 150 ± 2 | 332 ± 26 | 129 ± 2 | 310 ± 21 | 149 ± 26 |
| NBCoV37 | 311 ± 33 | 130 ± 35 | 42.5 ± 1 | 215 ± 10 | 73 ± 14 |
| NBCoV63 | 42.5 ± 1 | 105 ± 1 | 60 ± 3 | 26 ± 0.5 | 45.5 ± 2 |
| SARS-CoV-1 | ||||
|---|---|---|---|---|
| Compound | 293T/ACE2 Cells | A549/ACE2/TMPRSS2 Cells | ||
| IC50 (nM) * | SI | IC50 (nM) * | SI | |
| NBCoV35 | 380 ± 69 | 132 | 363 ± 11 | 138 |
| NBCoV36 | 660 ± 30 | 65 | 147 ± 19 | 340 |
| NBCoV37 | 340 ± 34 | 125 | 95 ± 1 | 526 |
| NBCoV63 | 110 ± 0.5 | 455 | 59 ± 1.5 | 848 |
| Compound | MERS-CoV | |||||
|---|---|---|---|---|---|---|
| Caco-2 Cells | MRC-5 Cells | |||||
| IC50 (nM) a | CC50 (µM) a | SI | IC50 (nM) a | CC50 (µM) a | SI | |
| NBCoV35 | 472 ± 43 | >50 | >106 | 715 ± 43 | >50 | >70 |
| NBCoV36 | 428 ± 34 | >50 | >117 | 837 ± 38 | >50 | >60 |
| NBCoV37 | 161 ± 53 | >50 | >311 | 229 ± 20 | 48 ± 2 | 210 |
| NBCoV63 | 75 ± 25 | >50 | >667 | 102 ± 1 | >50 | >490 |
| Compounds | Calu-3 Cells | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SARS-CoV-2 | SARS-CoV-1 | MERS-CoV | Toxicity | ||||||
| USA-WA1/2020 | Omicron BA.4/BA.5 | ||||||||
| IC50 (nM) a | SI | IC50 (nM) a | SI | IC50 (nM) a | SI | IC50 (nM) a | SI | CC50 (µM) a | |
| NBCoV35 | 360 ± 35 | >139 | 520 ± 27 | >96 | 1247 ± 45 | >40 | 1667 ± 225 | >30 | >50 |
| NBCoV36 | 433 ± 49 | >116 | 565 ± 79 | >89 | 627 ± 58 | >80 | 589 ± 117 | >85 | >50 |
| NBCoV37 | 368 ± 45 | >136 | 583 ± 35 | >86 | 1000 ± 132 | >50 | 877 ± 50 | >57 | >50 |
| NBCoV63 | 120 ± 6 | 417 | 126 ± 5 | 397 | 133 ± 28 | 376 | 220 ± 30 | 227 | 50 |
| Compound | A-MLV IC50 (nM) a |
|---|---|
| NBCoV35 | 1825 ± 25 |
| NBCoV36 | 3300 ± 100 |
| NBCoV37 | 1511 ± 88 |
| NBCoV63 | 303 ± 29 |
| Virus | IC50 (µM) 2 | CC50 (µM) 3 | |
|---|---|---|---|
| NBCoV63 | Remdesivir (Control) | ||
| SARS-CoV-2 Hong Kong | 2.04 | 3.44 | >50 |
| SARS-CoV-2 Delta (B.1.617.2) | 9.27 | 12.5 | |
| SARS-CoV-2 Omicron (BA.2.12.1) | 5.45 | 2.82 | |
| SARS-CoV-1 Urbani | 3.98 | 1.08 | |
| MERS-CoV | 8.01 | 17.8 | |
| Assay Performed | In Vitro ADMET | Inhibitor |
|---|---|---|
| NBCoV63 | ||
| Solubility (µM) | Phosphate buffer, pH7.4 | 6.25 |
| Caco-2 permeability (mean Papp, ×10−6 cm/s) | A-to-B | 0.00242 |
| B-to-A | 0.19 | |
| Efflux Ratio | 42.3 | |
| Metabolic stability (human liver microsomes) | Clint (µL/min/mg protein) | 3.4 |
| Half-life (min) | >180 | |
| Protein binding (human plasma) | % bound | >99.5 |
| Cytochrome P450 inhibition, IC50 (µM) | CYP1A2 (a-Naphthoflavone) | >25 |
| CYP2B6 (Ticlopidine) | >25 | |
| CYP2C8 (Quercetin) | >25 | |
| CYP2C9 (Sulphaphenazole) | >25 | |
| CYP2C19 (Ticlopidine) | >25 | |
| CYP2D6 (Quinidine) | >25 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Curreli, F.; Chau, K.; Tran, T.-T.; Nicolau, I.; Ahmed, S.; Das, P.; Hillyer, C.D.; Premenko-Lanier, M.; Debnath, A.K. Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors. Viruses 2023, 15, 1001. https://doi.org/10.3390/v15041001
Curreli F, Chau K, Tran T-T, Nicolau I, Ahmed S, Das P, Hillyer CD, Premenko-Lanier M, Debnath AK. Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors. Viruses. 2023; 15(4):1001. https://doi.org/10.3390/v15041001
Chicago/Turabian StyleCurreli, Francesca, Kent Chau, Thanh-Thuy Tran, Isabella Nicolau, Shahad Ahmed, Pujita Das, Christopher D. Hillyer, Mary Premenko-Lanier, and Asim K. Debnath. 2023. "Discovery of Highly Potent Small Molecule Pan-Coronavirus Fusion Inhibitors" Viruses 15, no. 4: 1001. https://doi.org/10.3390/v15041001