
One Health Surveillance Highlights Circulation of Viruses with Zoonotic Potential in Bats, Pigs, and Humans in Viet Nam
 , ,
, ,  , , , and add
Show full author list
, , , and add
Show full author list
Abstract
:1. Introduction
2. Material and Methods
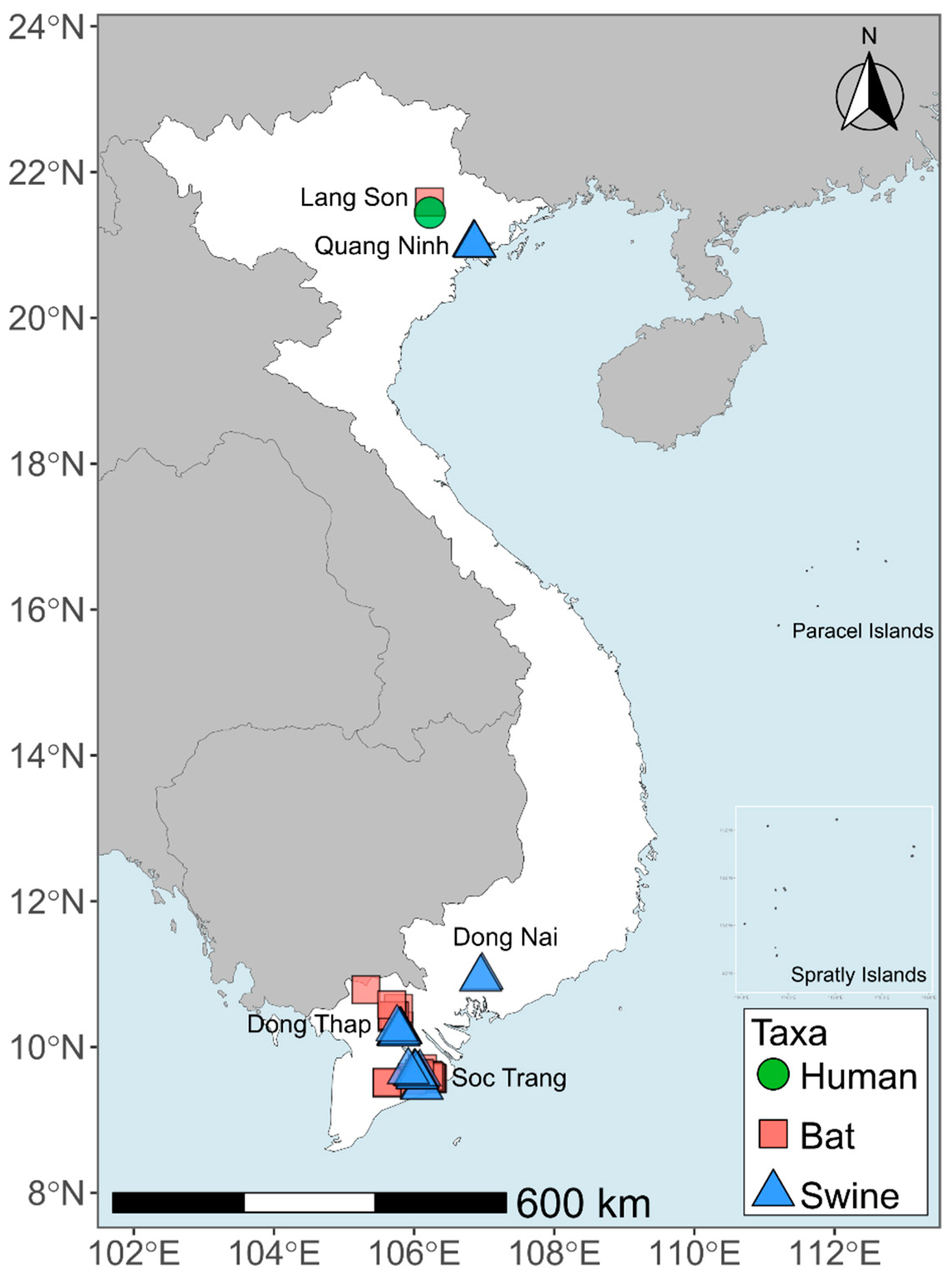
2.1. Sample Collection
2.2. Bat Species Identification
2.3. Viral Screening

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxa | Sample Type | CoVs | PmVs | Influenza | Flavi | Filo | Viral Species |
|---|---|---|---|---|---|---|---|
| Bats | Guano and feces | 32.7% (339/1035) a | 1.5% (13/835) | 0% (0/833) | 0% (0/835) | 0% (0/833) |
|
| Urine | 0% (0/55) | 16% (4/25) | 0% (0/25) | 0% (0/25) | 0% (0/25) |
| |
| Oral swabs | 10.5% (2/19) | 0% (0/14) | 0% (0/14) | 0% (0/14) | 0% (0/14) |
| |
| Rectal swabs | 18.2% (4/22) | 0% (0/17) | 0% (0/17) | 0% (0/17) | 0% (0/17) |
| |
| Total | 30.5% (345/1131) | 1.9% (17/888) | 0% (0/888) | 0% (0/888) | 0% (0/888) | ||
| Pigs | Nasal swabs | 0% (0/185) | 1.6% (3/185) | 8.1% (15/185) | 0% (0/185) | 0% (0/185) |
|
| Oral swabs | 40% (120/300) | 2.3% (7/300) | 3% (9/300) | 0% (0/300) | 0% (0/300) |
| |
| Total | 24.7% (120/485) | 2.1% (10/485) | 4.9% (24/485) | 0% (0/485) | 0% (0/485) | ||
| Humans | Whole blood | 0% (0/30) | 0% (0/30) | 0% (0/30) | 0% (0/30) | 0% (0/30) | |
| Oral swabs | 0% (0/30) | 0% (0/30) | 0% (0/30) | 0% (0/30) | 0% (0/30) | ||
| Total | 0% (0/60) | 0% (0/60) | 0% (0/60) | 0% (0/60) | 0% (0/60) |
2.4. Phylogenetic Analysis
2.5. Phylogeographic Analysis
2.6. Statistical Analysis of Seasonal Effect on CoV Shedding
2.7. Human Serology
3. Results
3.1. Virus Detection in Bat and Pig Samples
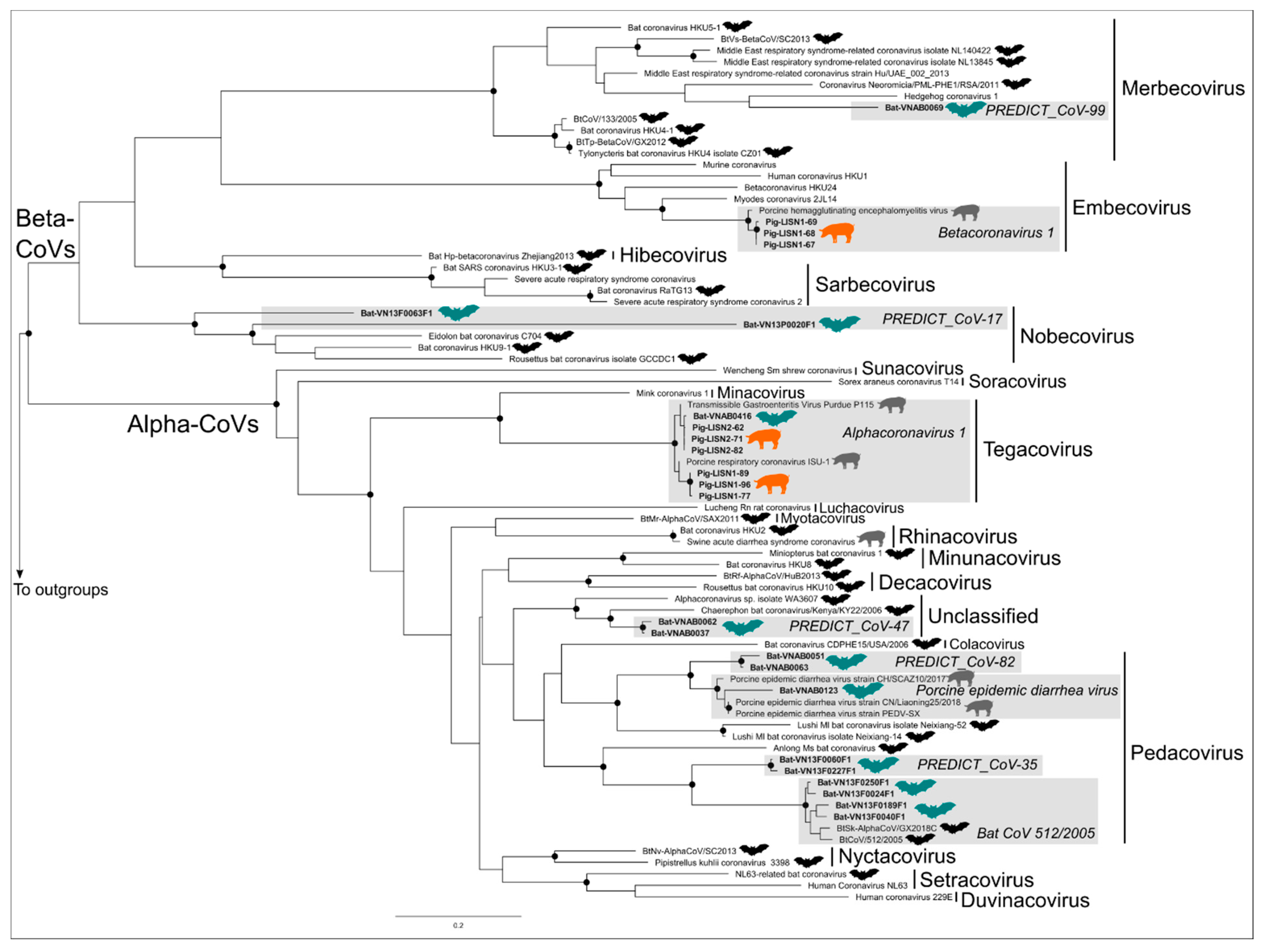
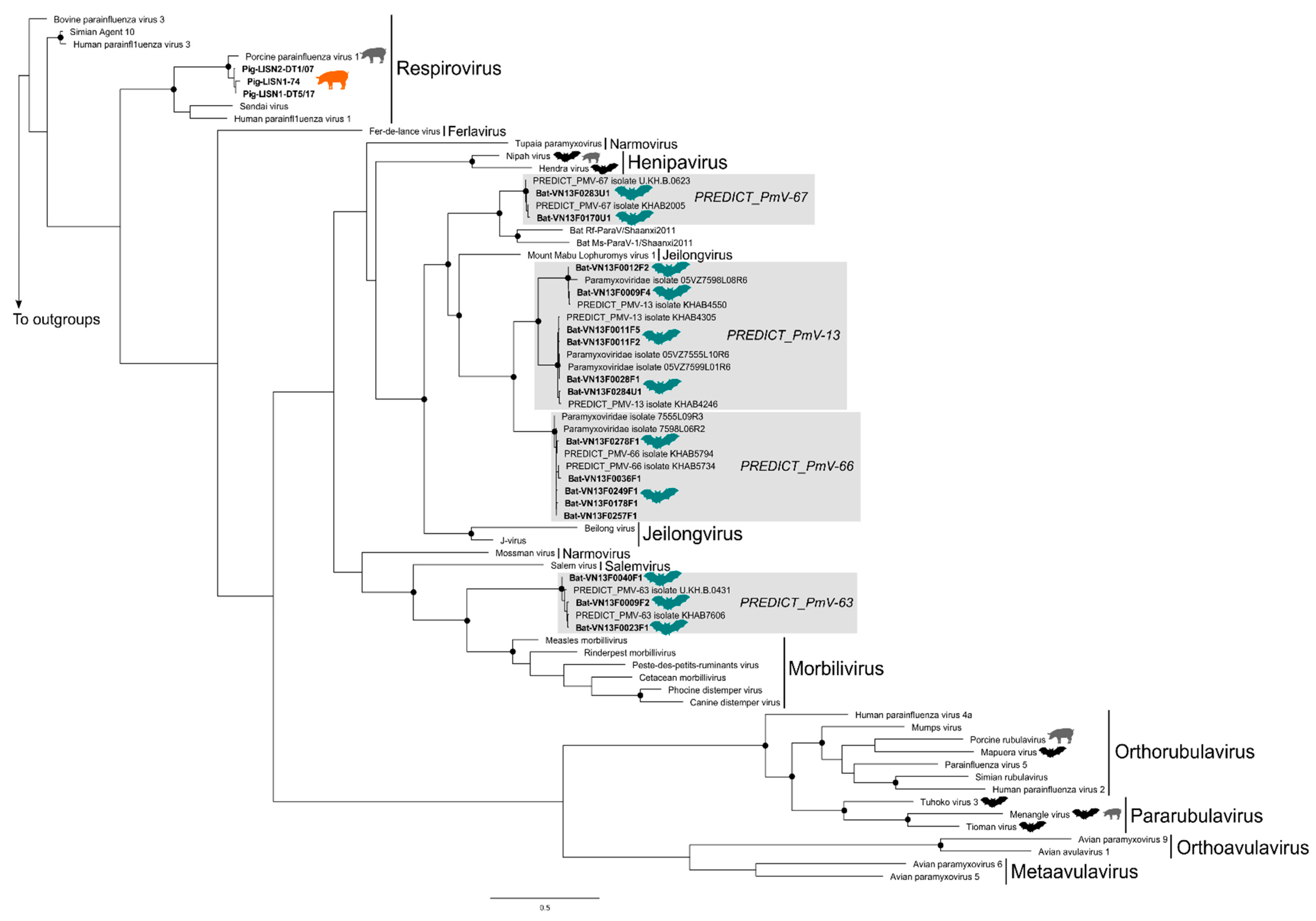
3.2. Phylogenetic Trees
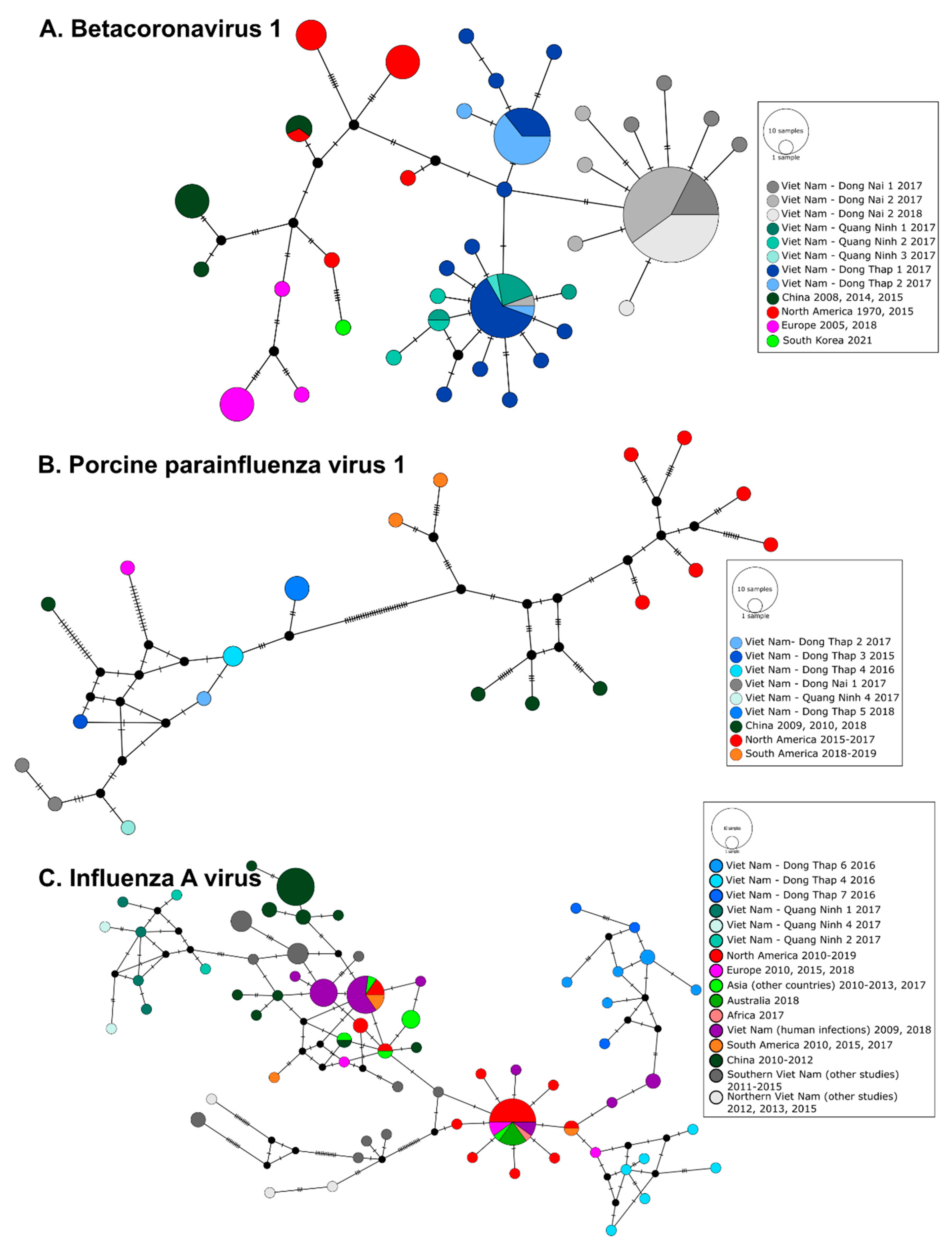
3.3. Phylogeography of Pig Viruses in Viet Nam
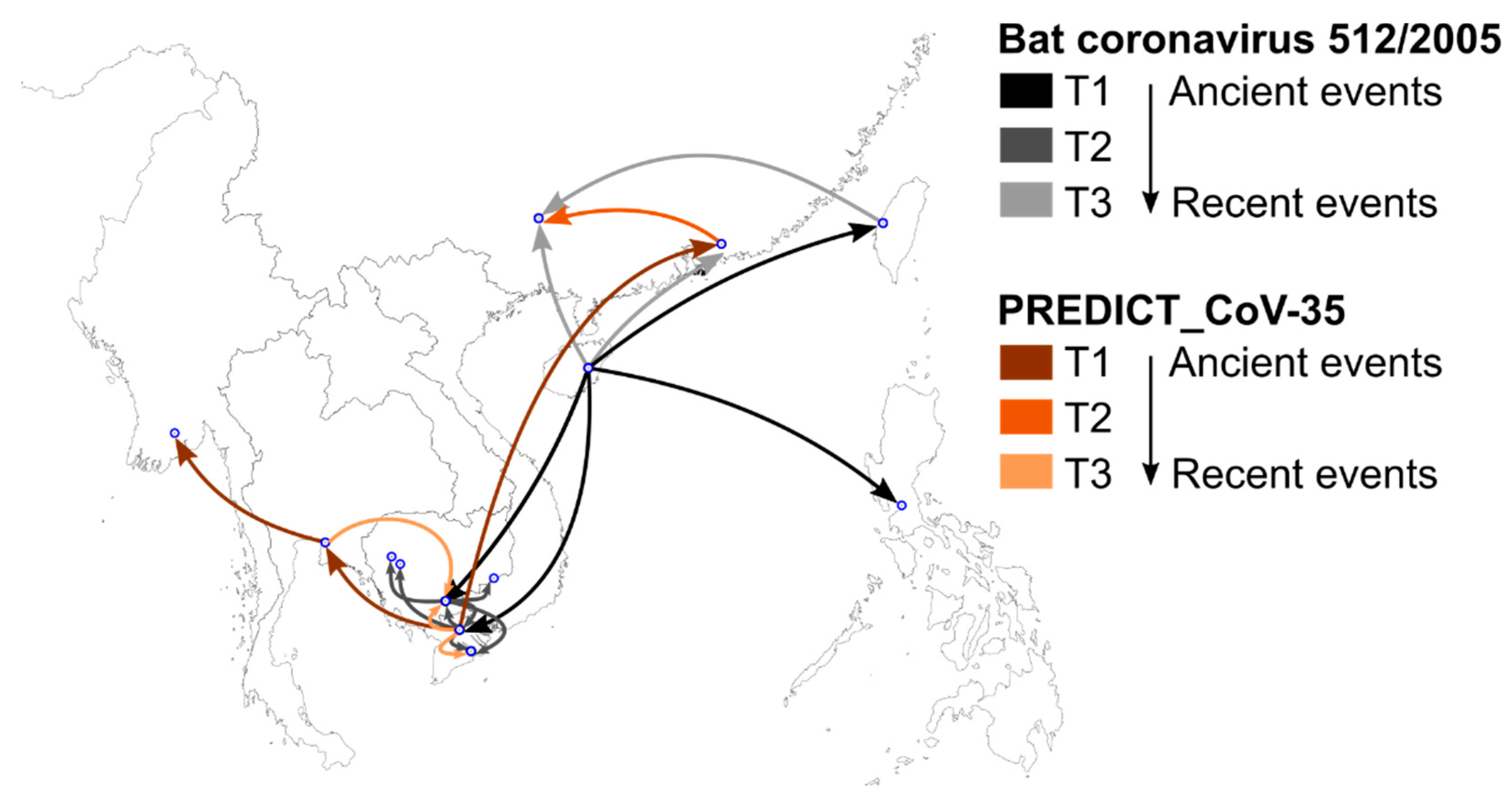
3.4. Phylogeography and Spatio-Temporal Spread of Bat CoVs
3.5. Seasonal Effect on CoV Detection in Bat Guano
3.6. Human Serology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Stärk, K.D.C.; Arroyo Kuribreña, M.; Dauphin, G.; Vokaty, S.; Ward, M.P.; Wieland, B.; Lindberg, A. One Health surveillance—More than a buzz word? Prev. Vet. Med. 2015, 120, 124–130. [Google Scholar] [CrossRef]
- World Bank. People, Pathogens and Our Planet: The Economics of One Health; World Bank: Washington, DC, USA, 2012. [Google Scholar]
- IPBES. Workshop Report on Biodiversity and Pandemics of the Intergovernmental Platform on Biodiversity and Ecosystem Services; Daszak, P., Amuasi, J., das Neves, C.G., Hayman, D., Kuiken, T., Roche, B., Zambrana-Torrelio, C., Buss, P., Dundarova, H., Feferholtz, Y., et al., Eds.; IPBES: Bonn, Germany, 2020. [Google Scholar]
- Aarestrup, F.M.; Bonten, M.; Koopmans, M. Pandemics—One Health preparedness for the next. Lancet Reg. Health—Eur. 2021, 9, 100210. [Google Scholar] [CrossRef] [PubMed]
- OHHLEP. One Health High-Level Expert Panel, Annual Report 2021; OHHLEP: Geneva, Switzerland, 2021. [Google Scholar]
- Machalaba, C.; Uhart, M.; Ryser-Degiorgis, M.P.; Karesh, W.B. Gaps in health security related to wildlife and environment affecting pandemic prevention and preparedness, 2007–2020. Bull. World Health Organ. 2021, 99, 342–350B. [Google Scholar] [CrossRef] [PubMed]
- Keusch, G.T.; Amuasi, J.H.; Anderson, D.E.; Daszak, P.; Eckerle, I.; Field, H.; Koopmans, M.; Lam, S.K.; Das Neves, C.G.; Peiris, M.; et al. Pandemic origins and a One Health approach to preparedness and prevention: Solutions based on SARS-CoV-2 and other RNA viruses. Proc. Natl. Acad. Sci. USA 2022, 119, e2202871119. [Google Scholar] [CrossRef] [PubMed]
- USDA. Livestock and Poultry: World Markets and Trade, April 2022. Available online: https://downloads.usda.library.cornell.edu/usda-esmis/files/73666448x/n5840036r/v979w6960/livestock_poultry.pdf (accessed on 17 March 2023).
- Dzung, N.M.; TuLiem, T. Pig Production and Marketing in Vietnam; National Institute of Animal Science: Hanoi, Viet Nam, 2015.
- Graham, R.L.; Baric, R.S. Recombination, Reservoirs, and the Modular Spike: Mechanisms of Coronavirus Cross-Species Transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Latinne, A.; Hu, B.; Olival, K.J.; Zhu, G.; Zhang, L.; Li, H.; Chmura, A.A.; Field, H.E.; Zambrana-Torrelio, C.; Epstein, J.H.; et al. Origin and cross-species transmission of bat coronaviruses in China. Nat. Commun. 2020, 11, 4235. [Google Scholar] [CrossRef] [PubMed]
- Leopardi, S.; Holmes, E.C.; Gastaldelli, M.; Tassoni, L.; Priori, P.; Scaravelli, D.; Zamperin, G.; De Benedictis, P. Interplay between co-divergence and cross-species transmission in the evolutionary history of bat coronaviruses. Infect. Genet. Evol. 2018, 58, 279–289. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Chapter Eight—Hosts and Sources of Endemic Human Coronaviruses. In Advances in Virus Research; Kielian, M., Mettenleiter, T., Roossinck, M., Eds.; Academic Press: Cambridge, MA, USA, 2018; pp. 163–188. [Google Scholar]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203.e3. [Google Scholar] [CrossRef]
- Corman, V.M.; Ithete, N.L.; Richards, L.R.; Schoeman, M.C.; Preiser, W.; Drosten, C.; Drexler, J.F. Rooting the Phylogenetic Tree of Middle East Respiratory Syndrome Coronavirus by Characterization of a Conspecific Virus from an African Bat. J. Virol. 2014, 88, 11297–11303. [Google Scholar] [CrossRef] [Green Version]
- Huynh, J.; Li, S.; Yount, B.; Smith, A.; Sturges, L.; Olsen, J.C.; Nagel, J.; Johnson, J.B.; Agnihothram, S.; Gates, J.E.; et al. Evidence Supporting a Zoonotic Origin of Human Coronavirus Strain NL63. J. Virol. 2012, 86, 12816–12825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corman, V.M.; Baldwin, H.J.; Tateno, A.F.; Zerbinati, R.M.; Annan, A.; Owusu, M.; Nkrumah, E.E.; Maganga, G.D.; Oppong, S.; Adu-Sarkodie, Y.; et al. Evidence for an Ancestral Association of Human Coronavirus 229E with Bats. J. Virol. 2015, 89, 11858–11870. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Corman, V.M.; Muller, M.A.; Maganga, G.D.; Vallo, P.; Binger, T.; Gloza-Rausch, F.; Cottontail, V.M.; Rasche, A.; Yordanov, S.; et al. Bats host major mammalian paramyxoviruses. Nat. Commun. 2012, 3, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latinne, A.; Morand, S. Climate Anomalies and Spillover of Bat-Borne Viral Diseases in the Asia-Pacific Region and the Arabian Peninsula. Viruses 2022, 14, 1100. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.A.; Smith, C.; Marsh, G.A.; Field, H.; Wang, L.-F. Evidence of bat origin for Menangle virus, a zoonotic paramyxovirus first isolated from diseased pigs. J. Gen. Virol. 2012, 93, 2590–2594. [Google Scholar] [CrossRef]
- Chua, K.B.; Bellini, W.J.; Rota, P.A.; Harcourt, B.H.; Tamin, A.; Lam, S.K.; Ksiazek, T.G.; Rollin, P.E.; Zaki, S.R.; Shieh, W.-J.; et al. Nipah Virus: A Recently Emergent Deadly Paramyxovirus. Science 2000, 288, 1432–1435. [Google Scholar] [CrossRef]
- Chua, K.B.; Lek Koh, C.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Enserink, M. New Virus Fingered in Malaysian Epidemic. Science 1999, 284, 407–410. [Google Scholar] [CrossRef]
- Chant, K.; Chan, R.; Smith, M.; Dwyer, D.E.; Kirkland, P. Probable human infection with a newly described virus in the family Paramyxoviridae. Emerg. Infect. Dis. 1998, 4, 273–275. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.-L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-K.; Pascua, P.N.Q.; Song, M.-S. Swine influenza viruses: An Asian perspective. Curr. Top. Microbiol. Immunol. 2013, 370, 147–172. [Google Scholar]
- Baudon, E.; Chu, D.K.W.; Tung, D.D.; Thi Nga, P.; Vu Mai Phuong, H.; Le Khanh Hang, N.; Thanh, L.T.; Thuy, N.T.; Khanh, N.C.; Mai, L.Q.; et al. Swine influenza viruses in Northern Vietnam in 2013–2014. Emerg. Microbes Infect. 2018, 7, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sothearen, T.; Furey, N.M.; Jurgens, J.A. Effect of bat guano on the growth of five economically important plant species. J. Trop. Agric. 2014, 52, 169–173. [Google Scholar]
- Furey, N.M.; Racey, P.A.; Ith, S.; Touch, V.; Cappelle, J. Reproductive Ecology of Wrinkle-Lipped Free-Tailed Bats Chaerephon plicatus (Buchannan, 1800) in Relation to Guano Production in Cambodia. Diversity 2018, 10, 91. [Google Scholar] [CrossRef] [Green Version]
- Huong, N.Q.; Nga, N.T.T.; Long, N.V.; Luu, B.D.; Latinne, A.; Pruvot, M.; Phuong, N.T.; Quang, L.T.V.; Hung, V.V.; Lan, N.T.; et al. Coronavirus testing indicates transmission risk increases along wildlife supply chains for human consumption in Viet Nam, 2013–2014. PLoS ONE 2020, 15, e0237129. [Google Scholar] [CrossRef]
- PREDICT Consortium. PREDICT Emerging Pandemic Threats Project. Dataset; USAID Development Data Library: Washington, DC, USA, 2021.
- Townzen, J.; Brower, A.; Judd, D. Identification of mosquito bloodmeals using mitochondrial cytochrome oxidase subunit I and cytochrome b gene sequences. Med. Vet. Entomol. 2008, 22, 386–393. [Google Scholar] [CrossRef]
- Quan, P.-L.; Firth, C.; Street, C.; Henriquez, J.A.; Petrosov, A.; Tashmukhamedova, A.; Hutchison, S.K.; Egholm, M.; Osinubi, M.O.V.; Niezgoda, M.; et al. Identification of a Severe Acute Respiratory Syndrome Coronavirus-Like Virus in a Leaf-Nosed Bat in Nigeria. mBio 2010, 1, e00208-10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat Coronaviruses and Experimental Infection of Bats, the Philippines. Emerg. Infect. Dis. 2010, 16, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Chern, S.-W.W.; Li, Y.; Pallansch, M.A.; Anderson, L.J. Sensitive and Broadly Reactive Reverse Transcription-PCR Assays to Detect Novel Paramyxoviruses. J. Clin. Microbiol. 2008, 46, 2652–2658. [Google Scholar] [CrossRef] [Green Version]
- Anthony, S.J.; St. Leger, J.A.; Pugliares, K.; Ip, H.S.; Chan, J.M.; Carpenter, Z.W.; Navarrete-Macias, I.; Sanchez-Leon, M.; Saliki, J.T.; Pedersen, J.; et al. Emergence of Fatal Avian Influenza in New England Harbor Seals. mBio 2012, 3, e00166-12. [Google Scholar] [CrossRef] [Green Version]
- Zhai, J.; Palacios, G.; Towner, J.S.; Jabado, O.; Kapoor, V.; Venter, M.; Grolla, A.; Briese, T.; Paweska, J.; Swanepoel, R.; et al. Rapid Molecular Strategy for Filovirus Detection and Characterization. J. Clin. Microbiol. 2007, 45, 224–226. [Google Scholar] [CrossRef] [Green Version]
- Moureau, G.; Temmam, S.; Gonzalez, J.P.; Charrel, R.N.; Grard, G.; de Lamballerie, X. A Real-Time RT-PCR Method for the Universal Detection and Identification of Flaviviruses. Vector-Borne Zoonotic Dis. 2007, 7, 467–478. [Google Scholar] [CrossRef]
- Lee, J.; Hughes, T.; Lee, M.-H.; Field, H.; Rovie-Ryan, J.J.; Sitam, F.T.; Sipangkui, S.; Nathan, S.K.S.S.; Ramirez, D.; Kumar, S.V.; et al. No Evidence of Coronaviruses or Other Potentially Zoonotic Viruses in Sunda pangolins (Manis javanica) Entering the Wildlife Trade via Malaysia. EcoHealth 2020, 17, 406–418. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Diagnostic Detection of 2019-nCoV by Real-Time RT-PCR, Protocol Version 2. 2020. Available online: https://www.who.int/docs/default-source/coronaviruse/protocol-v2-1.pdf?sfvrsn=a9ef618c_2 (accessed on 17 March 2023).
- Nga, N.T.T.; Latinne, A.; Thuy, H.B.; Long, N.V.; Ngoc, P.T.B.; Anh, N.T.L.; Thai, N.V.; Phuong, T.Q.; Thai, H.V.; Hai, L.K.; et al. Evidence of SARS-CoV-2 Related Coronaviruses Circulating in Sunda pangolins (Manis javanica) Confiscated From the Illegal Wildlife Trade in Viet Nam. Front. Public Health 2022, 10, 826116. [Google Scholar] [CrossRef]
- Anthony, S.J.; Johnson, C.K.; Greig, D.J.; Kramer, S.; Che, X.; Wells, H.; Hicks, A.L.; Joly, D.O.; Wolfe, N.D.; Daszak, P.; et al. Global patterns in coronavirus diversity. Virus Evol. 2017, 3, vex012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Bhartiy, S.K.; Elangovan, V. Reproductive behaviour of Asiatic lesser yellow bat, Scotophilus kuhlii (Chiroptera: Vespertilionidae) in Uttar Pradeh, India. J. Exp. Zool. India 2020, 23, 1577–1585. [Google Scholar]
- Chen, S.-F.; Huang, S.-S.; Lu, D.-J.; Shen, T.-J. Postnatal growth and age estimation in Scotophilus kuhlii. Zoo Biol. 2016, 35, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Thong, V.D. Taxonomic and distributional assessments of Chaerephon plicatus (Chiroptera: Molossidae) from Vietnam. Acad. J. Biol. 2015, 36, 479–486. [Google Scholar] [CrossRef] [Green Version]
- O’Hearn, A.E.; Voorhees, M.A.; Fetterer, D.P.; Wauquier, N.; Coomber, M.R.; Bangura, J.; Fair, J.N.; Gonzalez, J.-P.; Schoepp, R.J. Serosurveillance of viral pathogens circulating in West Africa. Virol. J. 2016, 13, 163. [Google Scholar] [CrossRef] [Green Version]
- Ricks, K.M.; Koehler, J.; Shoemaker, C.; Voorhees, M.; Schoepp, R. Development of a sustainable diagnostic toolbox for serosurveillance of West African infectious diseases. Int. J. Infect. Dis. 2019, 79, 24–25. [Google Scholar] [CrossRef] [Green Version]
- Ricks, K.M.; Shoemaker, C.J.; Dupuy, L.C.; Flusin, O.; Voorhees, M.A.; Fulmer, A.N.; Badger, C.V.; Schmaljohn, C.S.; Schoepp, R.J. Development of a bead-based immunoassay using virus-like particles for detection of alphaviral humoral response. J. Virol. Methods 2019, 270, 12–17. [Google Scholar] [CrossRef]
- Takemae, N.; Harada, M.; Nguyen Phuong, T.; Nguyen, T.; Nguyen Tien, N.; To Thanh, L.; Nguyen Tho, D.; Pham Vu, P.; Le Vu, T.; Do Hoa, T.; et al. Influenza A Viruses of Swine (IAV-S) in Vietnam from 2010 to 2015: Multiple Introductions of A(H1N1)pdm09 Viruses into the Pig Population and Diversifying Genetic Constellations of Enzootic IAV-S. J. Virol. 2017, 91, e01490-16. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Lacroix, A.; Duong, V.; Hul, V.; San, S.; Davun, H.; Omaliss, K.; Chea, S.; Hassanin, A.; Theppangna, W.; Silithammavong, S.; et al. Genetic diversity of coronaviruses in bats in Lao PDR and Cambodia. Infect. Genet. Evol. 2017, 48, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Valitutto, M.T.; Aung, O.; Tun, K.Y.N.; Vodzak, M.E.; Zimmerman, D.; Yu, J.H.; Win, Y.T.; Maw, M.T.; Thein, W.Z.; Win, H.H.; et al. Detection of novel coronaviruses in bats in Myanmar. PLoS ONE 2020, 15, e0230802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berto, A.; Anh, P.H.; Carrique-Mas, J.J.; Simmonds, P.; Van Cuong, N.; Tue, N.T.; Van Dung, N.; Woolhouse, M.E.; Smith, I.; Marsh, G.A.; et al. Detection of potentially novel paramyxovirus and coronavirus viral RNA in bats and rats in the Mekong Delta region of southern Viet Nam. Zoonoses Public Health 2018, 65, 30–42. [Google Scholar] [CrossRef] [Green Version]
- Grange, Z.L.; Goldstein, T.; Johnson, C.K.; Anthony, S.; Gilardi, K.; Daszak, P.; Olival, K.J.; O’Rourke, T.; Murray, S.; Olson, S.H.; et al. Ranking the risk of animal-to-human spillover for newly discovered viruses. Proc. Natl. Acad. Sci. USA 2021, 118, e2002324118. [Google Scholar] [CrossRef]
- Afelt, A.; Lacroix, A.; Zawadzka-Pawlewska, U.; Pokojski, W.; Buchy, P.; Frutos, R. Distribution of bat-borne viruses and environment patterns. Infect. Genet. Evol. 2018, 58, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617. [Google Scholar] [CrossRef]
- Huang, Y.-W.; Dickerman, A.W.; Piñeyro, P.; Li, L.; Fang, L.; Kiehne, R.; Opriessnig, T.; Meng, X.-J. Origin, Evolution, and Genotyping of Emergent Porcine Epidemic Diarrhea Virus Strains in the United States. mBio 2013, 4, e00737-13. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Vlasova, A.N.; Kenney, S.P.; Saif, L.J. Emerging and re-emerging coronaviruses in pigs. Curr. Opin. Virol. 2019, 34, 39–49. [Google Scholar] [CrossRef]
- Mai, T.N.; Yamazaki, W.; Bui, T.P.; Nguyen, V.G.; Le Huynh, T.M.; Mitoma, S.; Daous, H.E.; Kabali, E.; Norimine, J.; Sekiguchi, S. A descriptive survey of porcine epidemic diarrhea in pig populations in northern Vietnam. Trop. Anim. Health Prod. 2020, 52, 3781–3788. [Google Scholar] [CrossRef]
- Baudon, E.; Fournié, G.; Hiep, D.T.; Pham, T.T.H.; Duboz, R.; Gély, M.; Peiris, M.; Cowling, B.J.; Ton, V.D.; Peyre, M. Analysis of Swine Movements in a Province in Northern Vietnam and Application in the Design of Surveillance Strategies for Infectious Diseases. Transbound. Emerg. Dis. 2017, 64, 411–424. [Google Scholar] [CrossRef] [Green Version]
- Cappelle, J.; Furey, N.; Hoem, T.; Ou, T.P.; Lim, T.; Hul, V.; Heng, O.; Chevalier, V.; Dussart, P.; Duong, V. Longitudinal monitoring in Cambodia suggests higher circulation of alpha and betacoronaviruses in juvenile and immature bats of three species. Sci. Rep. 2021, 11, 24145. [Google Scholar] [CrossRef]
- Montecino-Latorre, D.; Goldstein, T.; Gilardi, K.; Wolking, D.; Van Wormer, E.; Kazwala, R.; Ssebide, B.; Nziza, J.; Sijali, Z.; Cranfield, M.; et al. Reproduction of East-African bats may guide risk mitigation for coronavirus spillover. One Health Outlook 2020, 2, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, L.E.; Fagre, A.C.; Chen, B.; Carlson, C.J.; Becker, D.J. Sampling strategies and pre-pandemic surveillance gaps for bat coronaviruses. bioRxiv, 2022; preprint. [Google Scholar]
- WHO. Dengue in Viet Nam. 2023. Available online: https://www.who.int/vietnam/health-topics/dengue (accessed on 17 March 2023).
- ECDC. Dengue Worldwide Overview. 2023. Available online: https://www.ecdc.europa.eu/en/dengue-monthly#:~:text=The%20five%20countries%20reporting%20most%20new%20deaths%20are%20Indonesia%20(229,have%20been%20reported%20in%20France (accessed on 17 March 2023).
- CDC. Areas at Risk for Chikungunya. 2022. Available online: https://www.cdc.gov/chikungunya/geo/index.html (accessed on 17 March 2023).
- Sulkin, S.E.; Allen, R.; Miura, T.; Toyokawa, K. Studies of arthropod-borne virus infections in chiroptera. VI. Isolation of Japanese B encephalitis virus from naturally infected bats. Am. J. Trop. Med. Hyg. 1970, 19, 77–87. [Google Scholar] [CrossRef]
- Wang, J.-L.; Pan, X.-L.; Zhang, H.-L.; Fu, S.-H.; Wang, H.-Y.; Tang, Q.; Wang, L.-F.; Liang, G.-D. Japanese encephalitis viruses from bats in Yunnan, China. Emerg. Infect. Dis. 2009, 15, 939. [Google Scholar] [CrossRef] [PubMed]
- Weissenböck, H.; Kolodziejek, J.; Url, A.; Lussy, H.; Rebel-Bauder, B.; Nowotny, N. Emergence of Usutu virus, an African mosquito-borne flavivirus of the Japanese encephalitis virus group, central Europe. Emerg. Infect. Dis. 2022, 8, 652. [Google Scholar] [CrossRef]
- Cadar, D.; Becker, N.; Campos Rde, M.; Börstler, J.; Jöst, H.; Schmidt-Chanasit, J. Usutu virus in bats, Germany, 2013. Emerg. Infect. Dis. 2014, 20, 1771–1773. [Google Scholar] [CrossRef]
- Jeffrey Root, J. West Nile virus associations in wild mammals: A synthesis. Arch. Virol. 2013, 158, 735–752. [Google Scholar] [CrossRef]






| Guano Farms in Southern Viet Nam | ||||
|---|---|---|---|---|
| Month (Year) | Season | Samples Tested | Samples Positive | Percentage Positive |
| January (2013) | Dry; no birth | 8 | 1 | 12.5% |
| June (2018) | Wet; birth + lactation | 130 | 15 | 11.5% |
| July (2017) | Wet; birth + lactation | 200 | 43 | 21.5% |
| October (2013) | Wet; no birth | 305 | 233 | 76.4% |
| December (2017) | Dry; no birth | 105 | 31 | 29.5% |
| Bat cave in northern Viet Nam | ||||
| Month (Year) | Season | Samples Tested | Samples Positive | Percentage Positive |
| April (2017) | Dry; birth | 74 | 9 | 12.2% |
| July (2017) | Wet; no birth | 126 | 7 | 5.5% |
| September (2018) | Wet; no birth | 100 | 0 | 0% |
| Target Virus | IgG | IgM |
|---|---|---|
| Positive/Suspect/Total (% Positive) | Positive/Suspect/Total (% Positive) | |
| Alphaviruses (CHIKV) | 0/1/30 | 0/1/30 |
| Crimean–Congo hemorrhagic fever virus (CCHFV) | 1/5/30 (3.3 %) | 0/0/30 |
| Flaviviruses (DENV2) | 26/3/30 (86.7 %) | 0/0/30 |
| Hantaviruses (HNTV) | 0/5/30 | 0/0/30 |
| Lassa fever virus (LASV) | 0/2/30 | 0/2/30 |
| Rift Valley fever virus (RVFV) | 0/4/30 | 0/1/30 |
| Ebolaviruses (EBOV) | 0/3/30 | NA |
| Marburg virus (MARV) | 1/0/30 (3.3 %) | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latinne, A.; Nga, N.T.T.; Long, N.V.; Ngoc, P.T.B.; Thuy, H.B.; PREDICT Consortium; Long, N.V.; Long, P.T.; Phuong, N.T.; Quang, L.T.V.; et al. One Health Surveillance Highlights Circulation of Viruses with Zoonotic Potential in Bats, Pigs, and Humans in Viet Nam. Viruses 2023, 15, 790. https://doi.org/10.3390/v15030790
Latinne A, Nga NTT, Long NV, Ngoc PTB, Thuy HB, PREDICT Consortium, Long NV, Long PT, Phuong NT, Quang LTV, et al. One Health Surveillance Highlights Circulation of Viruses with Zoonotic Potential in Bats, Pigs, and Humans in Viet Nam. Viruses. 2023; 15(3):790. https://doi.org/10.3390/v15030790
Chicago/Turabian StyleLatinne, Alice, Nguyen Thi Thanh Nga, Nguyen Van Long, Pham Thi Bich Ngoc, Hoang Bich Thuy, PREDICT Consortium, Nguyen Van Long, Pham Thanh Long, Nguyen Thanh Phuong, Le Tin Vinh Quang, and et al. 2023. "One Health Surveillance Highlights Circulation of Viruses with Zoonotic Potential in Bats, Pigs, and Humans in Viet Nam" Viruses 15, no. 3: 790. https://doi.org/10.3390/v15030790