
Opportunities for CAR-T Cell Immunotherapy in HIV Cure
 , and
, and
Abstract
:1. Introduction
2. Development of the CAR Technology
2.1. Early Studies of CAR-T Cells
2.2. New Generations of CARs-T Cells
2.3. Translation of the CAR-T Cell Technology to the Clinic
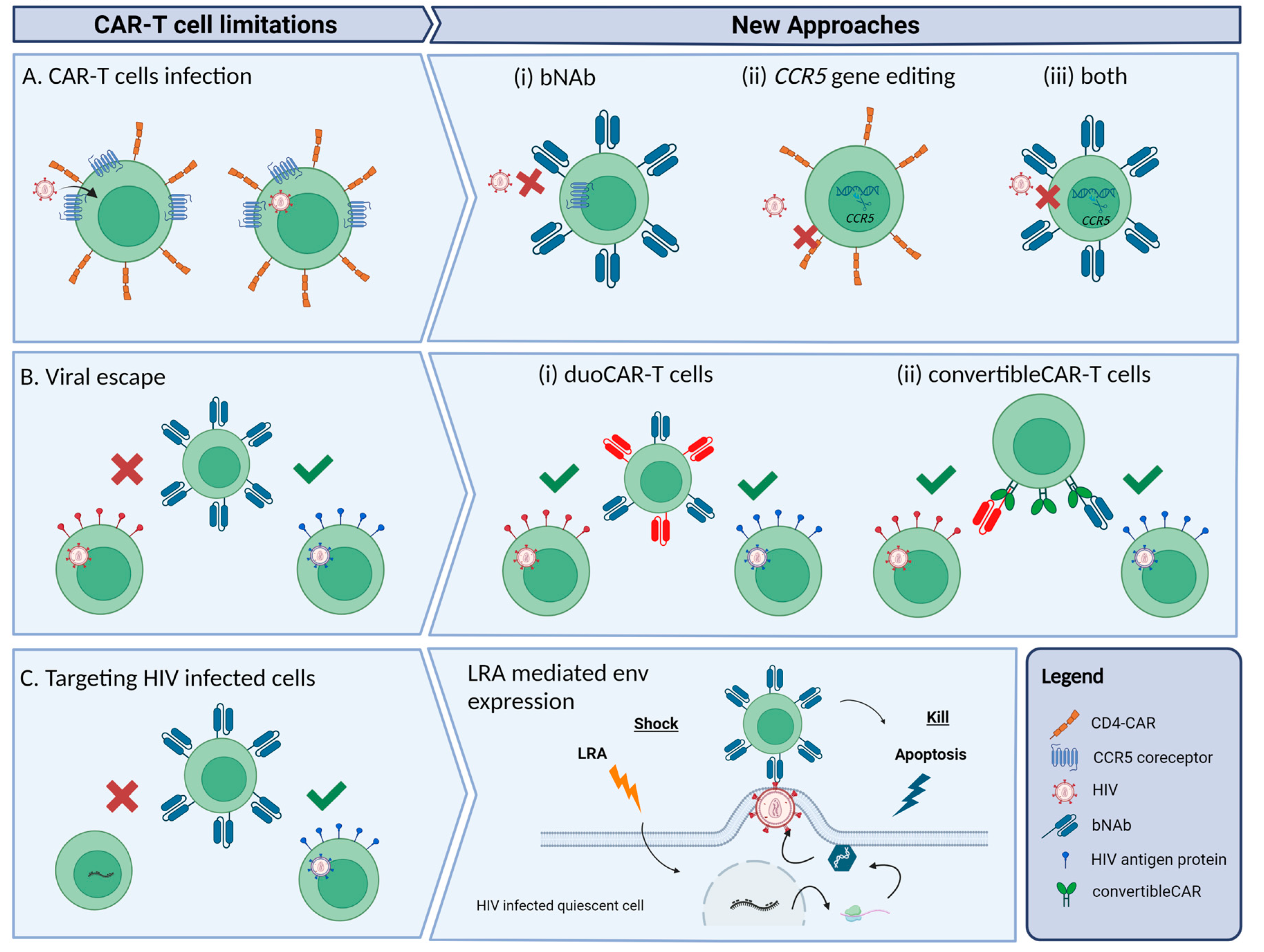
3. Difficulties and New Strategies to Achieve HIV Cure with CAR-T Cells
3.1. Avoiding CAR-T Cell Infection
3.2. Reducing Viral Escape
3.3. Targeting HIV Infected Cells
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Staszewski, S.; Miller, V.; Rehmet, S.; Stark, T.; De Crée, J.; De Brabander, M.; Peeters, M.; Andries, K.; Moeremans, M.; De Raeymaeker, M.; et al. Virological and Immunological Analysis of a Triple Combination Pilot Study with Loviride, Lamivudine and Zidovudine in HIV-1-Infected Patients. AIDS 1996, 10, F1–F7. [Google Scholar] [CrossRef]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In Vivo Fate of HIV-1-Infected T Cells: Quantitative Analysis of the Transition to Stable Latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent Infection of CD4+ T Cells Provides a Mechanism for Lifelong Persistence of HIV-1, Even in Patients on Effective Combination Therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Buzon, M.J.; Martin-Gayo, E.; Pereyra, F.; Ouyang, Z.; Sun, H.; Li, J.Z.; Piovoso, M.; Shaw, A.; Dalmau, J.; Zangger, N.; et al. Long-Term Antiretroviral Treatment Initiated at Primary HIV-1 Infection Affects the Size, Composition, and Decay Kinetics of the Reservoir of HIV-1-Infected CD4 T Cells. J. Virol. 2014, 88, 10056–10065. [Google Scholar] [CrossRef] [Green Version]
- Izopet, J.; Salama, G.; Pasquier, C.; Sandres, K.; Marchou, B.; Massip, P.; Puel, J. Decay of HIV-1 DNA in Patients Receiving Suppressive Antiretroviral Therapy. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 19, 478–483. [Google Scholar] [CrossRef]
- Parisi, S.G.; Andreis, S.; Mengoli, C.; Scaggiante, R.; Ferretto, R.; Manfrin, V.; Cruciani, M.; Giobbia, M.; Boldrin, C.; Basso, M.; et al. Baseline Cellular HIV DNA Load Predicts HIV DNA Decline and Residual HIV Plasma Levels during Effective Antiretroviral Therapy. J. Clin. Microbiol. 2012, 50, 258–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ananworanich, J.; Chomont, N.; Eller, L.A.; Kroon, E.; Tovanabutra, S.; Bose, M.; Nau, M.; Fletcher, J.L.K.; Tipsuk, S.; Vandergeeten, C.; et al. HIV DNA Set Point Is Rapidly Established in Acute HIV Infection and Dramatically Reduced by Early ART. EBioMedicine 2016, 11, 68–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casado, C.; Galvez, C.; Pernas, M.; Tarancon-Diez, L.; Rodriguez, C.; Sanchez-Merino, V.; Vera, M.; Olivares, I.; De Pablo-Bernal, R.; Merino-Mansilla, A.; et al. Permanent Control of HIV-1 Pathogenesis in Exceptional Elite Controllers: A Model of Spontaneous Cure. Sci. Rep. 2020, 10, 1902. [Google Scholar] [CrossRef] [Green Version]
- Mendoza, D.; Johnson, S.A.; Peterson, B.A.; Natarajan, V.; Salgado, M.; Dewar, R.L.; Burbelo, P.D.; Doria-Rose, N.A.; Graf, E.H.; Greenwald, J.H.; et al. Comprehensive Analysis of Unique Cases with Extraordinary Control over HIV Replication. Blood 2012, 119, 4645–4655. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.R.; Brennan, T.P.; O’Connell, K.A.; Siliciano, R.F.; Blankson, J.N. Evidence of CD8+ T-Cell-Mediated Selective Pressure on Human Immunodeficiency Virus Type 1 Nef in HLA-B*57+ Elite Suppressors. J. Virol. 2009, 83, 88–97. [Google Scholar] [CrossRef] [Green Version]
- Pereyra, F.; Palmer, S.; Miura, T.; Block, B.L.; Wiegand, A.; Rothchild, A.C.; Baker, B.; Rosenberg, R.; Cutrell, E.; Seaman, M.S.; et al. Persistent Low-Level Viremia in HIV-1 Elite Controllers and Relationship to Immunologic Parameters. J. Infect. Dis. 2009, 200, 984–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sáez-Cirión, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-Treatment HIV-1 Controllers with a Long-Term Virological Remission after the Interruption of Early Initiated Antiretroviral Therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef] [PubMed]
- Gálvez, C.; Urrea, V.; Dalmau, J.; Jimenez, M.; Clotet, B.; Monceaux, V.; Huot, N.; Leal, L.; González-Soler, V.; González-Cao, M.; et al. Extremely Low Viral Reservoir in Treated Chronically HIV-1-Infected Individuals. EBioMedicine 2020, 57, 102830. [Google Scholar] [CrossRef]
- Gálvez, C.; Urrea, V.; Garcia-Guerrero, M.D.C.; Bernal, S.; Benet, S.; Mothe, B.; Bailón, L.; Dalmau, J.; Martinez, A.; Nieto, A.; et al. Altered T-Cell Subset Distribution in the Viral Reservoir in HIV-1-Infected Individuals with Extremely Low Proviral DNA (LoViReTs). J. Intern. Med. 2022, 292, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Shergill, A.K.; Ho, T.; Killian, M.; Girling, V.; Epling, L.; Li, P.; Wong, L.K.; Crouch, P.; Deeks, S.G.; et al. The Distribution of HIV DNA and RNA in Cell Subsets Differs in Gut and Blood of HIV-Positive Patients on ART: Implications for Viral Persistence. J. Infect. Dis. 2013, 208, 1212–1220. [Google Scholar] [CrossRef] [Green Version]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. HIV-1 Remission Following CCR5Δ32/Δ32 Haematopoietic Stem Cell Transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.; Knops, E.; Lübke, N.; Wensing, A.M.; Martinez-Picado, J.; Kaiser, R.; Nijhuis, M.; Salgado, M.; Harrer, T.; Heger, E.; et al. Analytic Treatment Interruption (ATI) after Allogeneic CCR5-D32 HSCT in 2013. In Proceedings of the CROI, Seattle, WA, USA, 4–7 March 2019. [Google Scholar]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-Term Control of HIV by CCR5 Delta32/Delta32 Stem-Cell Transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Henrich, T.J.; Hanhauser, E.; Marty, F.M.; Sirignano, M.N.; Keating, S.; Lee, T.H.; Robles, Y.P.; Davis, B.T.; Li, J.Z.; Heisey, A.; et al. Antiretroviral-Free HIV-1 Remission and Viral Rebound after Allogeneic Stem Cell Transplantation: Report of 2 Cases. Ann. Intern. Med. 2014, 161, 319–327. [Google Scholar] [CrossRef]
- Salgado, M.; Kwon, M.; Gálvez, C.; Badiola, J.; Nijhuis, M.; Bandera, A.; Balsalobre, P.; Miralles, P.; Buño, I.; Martinez-Laperche, C.; et al. Mechanisms That Contribute to a Profound Reduction of the HIV-1 Reservoir after Allogeneic Stem Cell Transplant. Ann. Intern. Med. 2018, 169, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Bailón, L.; Llano, A.; Cedeño, S.; Escribà, T.; Rosás-Umbert, M.; Parera, M.; Casadellà, M.; Lopez, M.; Pérez, F.; Oriol-Tordera, B.; et al. Safety, Immunogenicity and Effect on Viral Rebound of HTI Vaccines in Early Treated HIV-1 Infection: A Randomized, Placebo-Controlled Phase 1 Trial. Nat. Med. 2022, 28, 2611–2621. [Google Scholar] [CrossRef]
- Blanch-Lombarte, O.; Gálvez, C.; Revollo, B.; Jiménez-Moyano, E.; Llibre, J.M.; Manzano, J.L.; Boada, A.; Dalmau, J.; Speiser, D.E.; Clotet, B.; et al. Enhancement of Antiviral CD8+ T-Cell Responses and Complete Remission of Metastatic Melanoma in an HIV-1-Infected Subject Treated with Pembrolizumab. J. Clin. Med. 2019, 8, 2089. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of Immunoglobulin-T-Cell Receptor Chimeric Molecules as Functional Receptors with Antibody-Type Specificity (Chimeric Genes/Antibody Variable Region). Immunology. 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [Green Version]
- Walker, R.E.; Bechtel, C.M.; Natarajan, V. Long-Term in Vivo Survival of Receptor-Modified Syngeneic T Cells in Patients with Human Immunodeficiency Virus Infection. Blood 2000, 96, 467–474. [Google Scholar] [PubMed]
- Masiero, S.; Del Vecchio, C.; Gavioli, R.; Mattiuzzo, G.; Cusi, M.G.; Micheli, L.; Gennari, F.; Siccardi, A.; Marasco, W.A.; Palù, G.; et al. T-Cell Engineering by a Chimeric T-Cell Receptor with Antibody-Type Specificity for the HIV-1 Gp120. Gene Ther. 2005, 12, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, M.R.; Qin, L.; Zhang, D. Targeting of Human Immunodeficiency Virus-Infected Cells by CDS+ T Lymphocytes Armed With Universal T-Cell Receptors. Blood 1994, 84, 2878–2889. [Google Scholar] [CrossRef] [Green Version]
- Yang, O.O.; Tran, A.-C.; Kalams, S.A.; Johnson, R.P.; Roberts, M.R.; Walker, B.D. Lysis of HIV-1-Infected Cells and Inhibition of Viral Replication by Universal Receptor T Cells. Proc. Natl. Acad. Sci. USA 1997, 94, 11478–11483. [Google Scholar] [CrossRef] [Green Version]
- Mitsuyasu, R.T.; Anton, P.A.; Deeks, S.G.; Scadden, D.T.; Connick, E.; Downs, M.T.; Bakker, A.; Roberts, M.R.; June, C.H.; Jalali, S.; et al. Prolonged Survival and Tissue Trafficking Following Adoptive Transfer of CD4ζ Gene-Modified Autologous CD4+ and CD8+ T Cells in Human Immunodeficiency Virus–Infected Subjects. Blood 2000, 96, 785–793. [Google Scholar] [CrossRef]
- Deeks, S.G.; Wagner, B.; Anton, P.A.; Mitsuyasu, R.T.; Scadden, D.T.; Huang, C.; Macken, C.; Richman, D.D.; Christopherson, C.; June, C.H.; et al. A Phase II Randomized Study of HIV-Specific T-Cell Gene Therapy in Subjects with Undetectable Plasma Viremia on Combination Antiretroviral Therapy. Mol. Ther. 2002, 5, 788–797. [Google Scholar] [CrossRef]
- Scholler, J.; Brady, T.L.; Binder-Scholl, G.; Hwang, W.-T.; Plesa, G.; Hege, K.M.; Vogel, A.N.; Kalos, M.; Riley, J.L.; Deeks, S.G.; et al. Decade-Long Safety and Function of Retroviral-Modified Chimeric Antigen Receptor T-Cells HHS Public Access. Sci. Transl. Med. 2012, 4, 132–153. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zhang, W.; Xia, B.; Jing, S.; Du, Y.; Zou, F.; Li, R.; Lu, L.; Chen, S.; Li, Y.; et al. Broadly Neutralizing Antibody-Derived CAR T Cells Reduce Viral Reservoir in Individuals Infected with HIV-1. J. Clin. Investig. 2021, 131, e150211. [Google Scholar] [CrossRef] [PubMed]
- Finney, H.M.; Lawson, A.D.; Bebbington, C.R. Chimeric Receptors Providing Both Primary and Costimulatory Signaling in T Cells from a Single Gene Product. J. Immunol. 1998, 161, 2791–2797. [Google Scholar] [CrossRef] [PubMed]
- Lawson, H.M.; Finney, A.N.; Akbar, A.D.G. Chain ζ CD137 in Series with Signals from the TCR CD28, Inducible Costimulator, CD134, and from with Chimeric Receptors: Costimulation Activation of Resting Human Primary T Cells. J. Immunol. Ref. 2022, 172, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Seif, M.; Einsele, H.; Löffler, J. CAR T Cells Beyond Cancer: Hope for Immunomodulatory Therapy of Infectious Diseases. Front. Immunol. 2019, 10, 2711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savoldo, B.; Ramos, C.A.; Liu, E.; Mims, M.P.; Keating, M.J.; Carrum, G.; Kamble, R.T.; Bollard, C.M.; Gee, A.P.; Mei, Z.; et al. CD28 Costimulation Improves Expansion and Persistence of Chimeric Antigen Receptor-Modified T Cells in Lymphoma Patients. J. Clin. Investig. 2011, 121, 1822–1826. [Google Scholar] [CrossRef] [Green Version]
- Chmielewski, M.; Hombach, A.A.; Abken, H. Of CARs and TRUCKs: Chimeric Antigen Receptor (CAR) T Cells Engineered with an Inducible Cytokine to Modulate the Tumor Stroma. Immunol. Rev. 2014, 257, 83–90. [Google Scholar] [CrossRef]
- Kagoya, Y.; Tanaka, S.; Guo, T.; Anczurowski, M.; Wang, C.H.; Saso, K.; Butler, M.O.; Minden, M.D.; Hirano, N. A Novel Chimeric Antigen Receptor Containing a JAK-STAT Signaling Domain Mediates Superior Antitumor Effects. Nat. Med. 2018, 24, 352–359. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [Green Version]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric Antigen Receptor-Modified T Cells for Acute Lymphoid Leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [Green Version]
- Brentjens, R.J.; Davila, M.L.; Riviere, I.; Park, J.; Wang, X.; Cowell, L.G.; Bartido, S.; Stefanski, J.; Taylor, C.; Olszewska, M.; et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci. Transl. Med. 2013, 5, 177. [Google Scholar] [CrossRef] [Green Version]
- Kochenderfer, J.N.; Dudley, M.E.; Feldman, S.A.; Wilson, W.H.; Spaner, D.E.; Maric, I.; Stetler-Stevenson, M.; Phan, G.Q.; Hughes, M.S.; Sherry, R.M.; et al. B-Cell Depletion and Remissions of Malignancy along with Cytokine-Associated Toxicity in a Clinical Trial of Anti-CD19 Chimeric-Antigen-Receptor-Transduced T Cells. Blood 2012, 119, 2709–2720. [Google Scholar] [CrossRef] [PubMed]
- Papadouli, I.; Mueller-Berghaus, J.; Beuneu, C.; Ali, S.; Hofner, B.; Petavy, F.; Tzogani, K.; Miermont, A.; Norga, K.; Kholmanskikh, O.; et al. EMA Review of Axicabtagene Ciloleucel (Yescarta) for the Treatment of Diffuse Large B-Cell Lymphoma. Oncologist 2020, 25, 894–902. [Google Scholar] [CrossRef]
- Alnefaie, A.; Albogami, S.; Asiri, Y.; Ahmad, T.; Alotaibi, S.S.; Al-Sanea, M.M.; Althobaiti, H. Chimeric Antigen Receptor T-Cells: An Overview of Concepts, Applications, Limitations, and Proposed Solutions. Front. Bioeng. Biotechnol. 2022, 10, 797440. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Maus, M.V.; Porter, D.L. Chimeric Antigen Receptor T Cell Therapy: 25years in the Making. Blood Rev. 2016, 30, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a Reservoir for HIV-1 in Patients on Highly Active Antiretroviral Therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Bitton, N.; Verrier, F.; Debré, P.; Gorochov, G. Characterization of T Cell-Expressed Chimeric Receptors with Antibody-Type Specificity for the CD4 Binding Site of HIV-1 Gp120. Eur. J. Immunol. 1998, 28, 4177–4187. [Google Scholar] [CrossRef]
- Zhen, A.; Kamata, M.; Rezek, V.; Rick, J.; Levin, B.; Kasparian, S.; Chen, I.S.; Yang, O.O.; Zack, J.A.; Kitchen, S.G. HIV-Specific Immunity Derived From Chimeric Antigen Receptor-Engineered Stem Cells. Mol. Ther. 2015, 23, 1358–1367. [Google Scholar] [CrossRef] [Green Version]
- York, J.; Gowrishankar, K.; Micklethwaite, K.; Palmer, S.; Cunningham, A.L.; Nasr, N. Evolving Strategies to Eliminate the CD4 T Cells HIV Viral Reservoir via CAR T Cell Immunotherapy. Front. Immunol. 2022, 13, 873301. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Mascola, J.R.; Nabel, G.J. Broadly Neutralizing Antibodies and the Search for an HIV-1 Vaccine: The End of the Beginning. Nat. Rev. Immunol. 2013, 13, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [Green Version]
- Rothemejer, F.H.; Lauritsen, N.P.; Juhl, A.K.; Schleimann, M.H.; König, S.; Søgaard, O.S.; Bak, R.O.; Tolstrup, M. Development of HIV-Resistant CAR T Cells by CRISPR/Cas-Mediated CAR Integration into the CCR5 Locus. Viruses 2023, 15, 202. [Google Scholar] [CrossRef]
- Huang, Y.; Paxton, W.A.; Wolinsky, S.M.; Neumann, A.U.; Zhang, L.; He, T.; Kang, S.; Ceradini, D.; Jin, Z.; Yazdanbakhsh, K.; et al. The Role of a Mutant CCR5 Allele in HIV–1 Transmission and Disease Progression. Nat. Med. 1996, 2, 1240–1243. [Google Scholar] [CrossRef] [PubMed]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hütter, G.; et al. Challenges in Detecting HIV Persistence during Potentially Curative Interventions: A Study of the Berlin Patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef] [PubMed]
- Jensen, B.-E.O.; Knops, E.; Cords, L.; Lübke, N.; Salgado, M.; Busman-Sahay, K.; Estes, J.D.; Huyveneers, L.E.P.; Perdomo-Celis, F.; Wittner, M.; et al. In-Depth Virological and Immunological Characterization of HIV-1 Cure after CCR5Δ32/Δ32 Allogeneic Hematopoietic Stem Cell Transplantation. Nat. Med. 2023, accepted. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tebas, P.; Jadlowsky, J.K.; Shaw, P.A.; Tian, L.; Esparza, E.; Brennan, A.L.; Kim, S.; Naing, S.Y.; Richardson, M.W.; Vogel, A.N.; et al. CCR5-Edited CD4+ T Cells Augment HIV-Specific Immunity to Enable Post-Rebound Control of HIV Replication. J. Clin. Investig. 2021, 131, e144486. [Google Scholar] [CrossRef]
- Rust, B.J.; Kean, L.S.; Colonna, L.; Brandenstein, K.E.; Poole, N.H.; Obenza, W.; Enstrom, M.R.; Maldini, C.R.; Ellis, G.I.; Fennessey, C.M.; et al. Robust Expansion of HIV CAR T Cells Following Antigen Boosting in ART-Suppressed Nonhuman Primates. Blood 2020, 15, 1722–1734. [Google Scholar] [CrossRef]
- Lichterfeld, M.; Kaufmann, D.E.; Yu, X.G.; Mui, S.K.; Addo, M.M.; Johnston, M.N.; Cohen, D.; Robbins, G.K.; Pae, E.; Alter, G.; et al. Loss of HIV-1-Specific CD8+ T Cell Proliferation after Acute HIV-1 Infection and Restoration by Vaccine-Induced HIV-1-Specific CD4+ T Cells. J. Exp. Med. 2004, 200, 701–712. [Google Scholar] [CrossRef]
- Maldini, C.R.; Gayout, K.; Leibman, R.S.; Dopkin, D.L.; Mills, J.P.; Shan, X.; Glover, J.A.; Riley, J.L. HIV-Resistant and HIV-Specific CAR-Modified CD4+ T Cells Mitigate HIV Disease Progression and Confer CD4+ T Cell Help In Vivo. Mol. Ther. 2020, 28, 1585–1599. [Google Scholar] [CrossRef]
- Lynch, R.M.; Boritz, E.; Coates, E.E.; DeZure, A.; Madden, P.; Costner, P.; Enama, M.E.; Plummer, S.; Holman, L.; Hendel, C.S.; et al. Virologic Effects of Broadly Neutralizing Antibody VRC01 Administration during Chronic HIV-1 Infection. Sci. Transl. Med. 2015, 7, 319ra206. [Google Scholar] [CrossRef] [Green Version]
- Riley, J.L.; Montaner, L.J. Cell-Mediated Immunity to Target the Persistent Human Immunodeficiency Virus Reservoir. J. Infect. Dis. 2017, 215, S160–S171. [Google Scholar] [CrossRef] [Green Version]
- Anthony-Gonda, K.; Bardhi, A.; Ray, A.; Flerin, N.; Li, M.; Chen, W.; Ochsenbauer, C.; Kappes, J.C.; Krueger, W.; Worden, A.; et al. Multispecific Anti-HIV DuoCAR-T Cells Display Broad in Vitro Antiviral Activity and Potent in Vivo Elimination of HIV-Infected Cells in a Humanized Mouse Model. Sci. Transl. Med. 2019, 11, eaav5685. [Google Scholar] [CrossRef]
- Anthony-Gonda, K.; Ray, A.; Su, H.; Wang, Y.; Xiong, Y.; Lee, D.; Block, A.; Chilunda, V.; Weiselberg, J.; Zemelko, L.; et al. In Vivo Killing of Primary HIV-Infected Cells by Peripheral-Injected Early Memory–Enriched Anti-HIV DuoCAR T Cells. JCI Insight 2022, 7, e161698. [Google Scholar] [CrossRef] [PubMed]
- Herzig, E.; Kim, K.C.; Packard, T.A.; Vardi, N.; Schwarzer, R.; Gramatica, A.; Deeks, S.G.; Williams, S.R.; Landgraf, K.; Killeen, N.; et al. Attacking Latent HIV with ConvertibleCAR-T Cells, a Highly Adaptable Killing Platform. Cell 2019, 179, 880–894.e10. [Google Scholar] [CrossRef] [PubMed]
- Bashiri, K.; Rezaei, N.; Nasi, M.; Cossarizza, A. The Role of Latency Reversal Agents in the Cure of HIV: A Review of Current Data. Immunol. Lett. 2018, 196, 135–139. [Google Scholar] [CrossRef]
- Liu, B.; Zou, F.; Lu, L.; Chen, C.; He, D.; Zhang, X.; Tang, X.; Liu, C.; Li, L.; Zhang, H. Chimeric Antigen Receptor T Cells Guided by the Single-Chain Fv of a Broadly Neutralizing Antibody Specifically and Effectively Eradicate Virus Reactivated from Latency in CD4+ T Lymphocytes Isolated from HIV-1-Infected Individuals Receiving Suppressive C. J. Virol. 2016, 90, 9712–9724. [Google Scholar] [CrossRef] [Green Version]
- Rasmussen, T.A.; Tolstrup, M.; Søgaard, O.S. Reversal of Latency as Part of a Cure for HIV-1. Trends Microbiol. 2016, 24, 90–97. [Google Scholar] [CrossRef]
- Walker-Sperling, V.E.; Pohlmeyer, C.W.; Tarwater, P.M.; Blankson, J.N. The Effect of Latency Reversal Agents on Primary CD8 + T Cells: Implications for Shock and Kill Strategies for Human Immunodeficiency Virus Eradication. EBioMedicine 2016, 8, 217–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.S.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-Competent Non-Induced Proviruses in the Latent Reservoir Increase Barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [Green Version]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV Reservoir Size and Persistence Are Driven by T Cell Survival and Homeostatic Proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Cantero-Pérez, J.; Grau-Expósito, J.; Serra-Peinado, C.; Rosero, D.A.; Luque-Ballesteros, L.; Astorga-Gamaza, A.; Castellví, J.; Sanhueza, T.; Tapia, G.; Lloveras, B.; et al. Resident Memory T Cells Are a Cellular Reservoir for HIV in the Cervical Mucosa. Nat. Commun. 2019, 10, 4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlenstiel, C.L.; Symonds, G.; Kent, S.J.; Kelleher, A.D. Block and Lock HIV Cure Strategies to Control the Latent Reservoir. Front. Cell. Infect. Microbiol. 2020, 10, 424. [Google Scholar] [CrossRef] [PubMed]
- Eberhard, J.M.; Angin, M.; Passaes, C.; Salgado, M.; Monceaux, V.; Knops, E.; Kobbe, G.; Jensen, B.; Christopeit, M.; Kröger, N.; et al. Vulnerability to Reservoir Reseeding Due to High Immune Activation after Allogeneic Hematopoietic Stem Cell Transplantation in Individuals with HIV-1. Sci. Transl. Med. 2020, 12, 542. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.W. SHIV Reservoirs Persist Following CAR T Cell-Mediated Depletion of B Cell Follicles in Nonhuman Primates. In Proceedings of the AIDS 2022, Montreal, QC, Canada, 29 July–2 August 2022. [Google Scholar]
- Ollerton, M.T.; Berger, E.A.; Connick, E.; Burton, G.F. HIV-1-Specific Chimeric Antigen Receptor T Cells Fail To Recognize and Eliminate the Follicular Dendritic Cell HIV Reservoir In Vitro. J. Virol. 2020, 94, e00190-20. [Google Scholar] [CrossRef]


{kind=link}
| Trial Registration | Study Title | Start Date | Phase | Country | CAR Generation | Type of CAR | Outcome | Reference |
|---|---|---|---|---|---|---|---|---|
| - | Prolonged survival and tissue trafficking following adoptive transfer of CD4ζ gene-modified autologous CD4+ and CD8+ T cells in human immunodeficiency virus–infected subjects | 1999 * | Phase II | USA | First | CD4ζ-based | Validation of the feasibility and antiviral activity | Mitsuyasu et al. Blood 2000. [28] |
| - | Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection | 1999 * | Phase I | USA | First | CD4ζ-based | Prove that administration is safe | Walker et al. Blood 2000. [24] |
| - | A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy | 2002 * | Phase II Randomized | USA | First | CD4ζ-based | Confirmed safety and feasibility, but no effect on HIV reservoirs | Deeks et al. Mol. Ther. 2002. [29] |
| NCT01013415 | A phase I/II study of the safety, survival, and trafficking of autologous CD4-ζ gene-modified T cells with and without extension Interleukin-2 in HIV infected patients | 2001 | Phase I Non-Randomized | USA | First | CD4ζ-based | Safety and long term persistence of modified T cells | Scholler et al. Sci. Trasl. Med. 2012. [30] |
| NCT03240328 | The effect of CAR-T cell therapy on the reconstitution of HIV-specific immune function | 2017 | Phase I | China | Third | bNAb-based | Long term in vivo persistence and no safety concerns | Liu et al. J. Clin. Invest. 2021. [31] |
| NCT03617198 | A pilot study of T cells genetically modified by Zinc Finger Nucleases SB-728mR and CD4 chimeric antigen receptor in HIV-infected subjects | 2019 | Phase I Randomized | USA | Second | CCR5 ZFN-treated CD4+ | Ongoing | - |
| NCT04648046 | Safety and anti-HIV activity of autologous CD4+ and CD8+ T cells transduced with a lentiviral vector encoding bi-specific anti-gp120 CAR molecules (LVgp120duoCAR-T) in anti-retroviral drug-treated HIV-1 infection | 2021 | Phase I/IIa Non-Randomized | USA | Second | CD4-based duoCAR | Ongoing | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campos-Gonzalez, G.; Martinez-Picado, J.; Velasco-Hernandez, T.; Salgado, M. Opportunities for CAR-T Cell Immunotherapy in HIV Cure. Viruses 2023, 15, 789. https://doi.org/10.3390/v15030789
Campos-Gonzalez G, Martinez-Picado J, Velasco-Hernandez T, Salgado M. Opportunities for CAR-T Cell Immunotherapy in HIV Cure. Viruses. 2023; 15(3):789. https://doi.org/10.3390/v15030789
Chicago/Turabian StyleCampos-Gonzalez, Gerard, Javier Martinez-Picado, Talia Velasco-Hernandez, and Maria Salgado. 2023. "Opportunities for CAR-T Cell Immunotherapy in HIV Cure" Viruses 15, no. 3: 789. https://doi.org/10.3390/v15030789