
Co-Detection of EBV and Human Polyomavirus JCPyV in a Case of AIDS-Related Multifocal Primary Central Nervous System Diffuse Large B-Cell Lymphoma


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
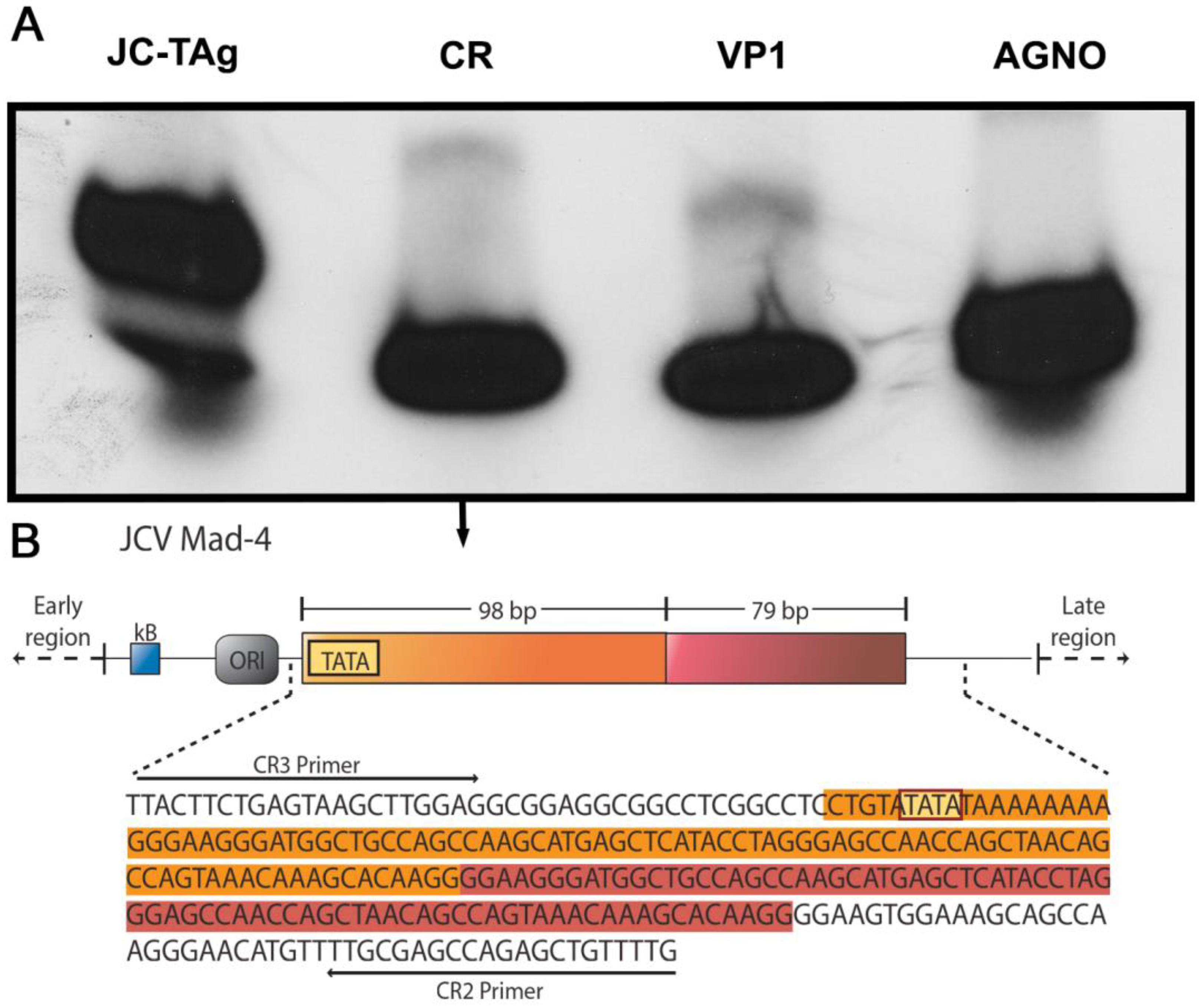{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Case Report
2.2. Immunohistochemistry
2.3. Double-Labeling Immunofluorescence
2.4. DNA Extraction and PCR Amplification
3. Results
3.1. Gross and Histopathological Aspects of the Tumor
3.2. Immunohistochemical Characterization of the Tumor
3.3. Expression of EBV and JCPyV Viral Proteins
3.4. Detection of JCPyV Genomic Sequences
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bailey, P. Intracranial sarcomatous tumors of leptomeningeal origin. JAMA Surg. 1929, 18, 1359–1402. [Google Scholar] [CrossRef]
- Henry, J.M.; Heffner, R.R.; Dillard, S.H.; Earle, K.M.; Davis, R.L. Primary malignant lymphomas of the central nervous system. Cancer 1974, 34, 1293–1302. [Google Scholar] [CrossRef] [PubMed]
- WHO Classification of Tumours Editorial Board (Ed.) Central Nervous System Tumours, 5th ed.; International Agency for Research on Cancer: Lyon, France, 2021. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; International Agency for Research on Cancer: Lyon, France, 2008; Volume 2. [Google Scholar]
- Kuker, W.; Nagele, T.; Korfel, A.; Heckl, S.; Thiel, E.; Bamberg, M.; Weller, M.; Herrlinger, U. Primary central nervous system lymphomas (PCNSL): MRI features at presentation in 100 patients. J. Neurooncol. 2005, 72, 169–177. [Google Scholar] [CrossRef]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenkier, T.N.; Blay, J.Y.; O’Neill, B.P.; Poortmans, P.; Thiel, E.; Jahnke, K.; Abrey, L.E.; Neuwelt, E.; Tsang, R.; Batchelor, T.; et al. Primary CNS lymphoma of T-cell origin: A descriptive analysis from the international primary CNS lymphoma collaborative group. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2005, 23, 2233–2239. [Google Scholar] [CrossRef]
- Shiels, M.S.; Pfeiffer, R.M.; Besson, C.; Clarke, C.A.; Morton, L.M.; Nogueira, L.; Pawlish, K.; Yanik, E.L.; Suneja, G.; Engels, E.A. Trends in primary central nervous system lymphoma incidence and survival in the U.S. Br. J. Haematol. 2016, 174, 417–424. [Google Scholar] [CrossRef] [Green Version]
- Rohilla, M.; Garg, S.; Bal, A.; Das, A.; Gupta, N.; Dey, P.; Srinivasan, R. Application of Hans Algorithm for Subcategorization of Diffuse Large B-Cell Lymphoma in Fine-Needle Aspiration Biopsy Cytology. Acta Cytol. 2022, 66, 14–22. [Google Scholar] [CrossRef]
- de Carvalho, P.S.; Leal, F.E.; Soares, M.A. Clinical and Molecular Properties of Human Immunodeficiency Virus-Related Diffuse Large B-Cell Lymphoma. Front. Oncol. 2021, 11, 675353. [Google Scholar] [CrossRef]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef]
- Ci, W.; Polo, J.M.; Cerchietti, L.; Shaknovich, R.; Wang, L.; Yang, S.N.; Ye, K.; Farinha, P.; Horsman, D.E.; Gascoyne, R.D.; et al. The BCL6 transcriptional program features repression of multiple oncogenes in primary B cells and is deregulated in DLBCL. Blood 2009, 113, 5536–5548. [Google Scholar] [CrossRef] [Green Version]
- Karube, K.; Campo, E. MYC alterations in diffuse large B-cell lymphomas. Semin. Hematol. 2015, 52, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Salaverria, I.; Philipp, C.; Oschlies, I.; Kohler, C.W.; Kreuz, M.; Szczepanowski, M.; Burkhardt, B.; Trautmann, H.; Gesk, S.; Andrusiewicz, M.; et al. Translocations activating IRF4 identify a subtype of germinal center-derived B-cell lymphoma affecting predominantly children and young adults. Blood 2011, 118, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Cattoretti, G.; Pasqualucci, L.; Ballon, G.; Tam, W.; Nandula, S.V.; Shen, Q.; Mo, T.; Murty, V.V.; Dalla-Favera, R. Deregulated BCL6 expression recapitulates the pathogenesis of human diffuse large B cell lymphomas in mice. Cancer Cell 2005, 7, 445–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iqbal, J.; Greiner, T.C.; Patel, K.; Dave, B.J.; Smith, L.; Ji, J.; Wright, G.; Sanger, W.G.; Pickering, D.L.; Jain, S.; et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia 2007, 21, 2332–2343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno, H. Pathogenetic and Clinical Implications of Non-Immunoglobulin; BCL6 Translocations in B-Cell Non-Hodgkin’s Lymphoma. J. Clin. Exp. Hematop. 2006, 46, 43–53. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Sola, D.; Victora, G.D.; Ying, C.Y.; Phan, R.T.; Saito, M.; Nussenzweig, M.C.; Dalla-Favera, R. The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat. Immunol. 2012, 13, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Papenhausen, P.; Shao, H. The Role of c-MYC in B-Cell Lymphomas: Diagnostic and Molecular Aspects. Genes 2017, 8, 116. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.; Nong, L.; Wang, W.; Liang, L.; Zheng, Y.; Liu, J.; Li, D.; Li, X.; Zhang, B.; Li, T. Prognostic impact of diffuse large B-cell lymphoma with extra copies of MYC, BCL2 and/or BCL6: Comparison with double/triple hit lymphoma and double expressor lymphoma. Diagn. Pathol. 2019, 14, 81. [Google Scholar] [CrossRef]
- Pillai, R.K.; Sathanoori, M.; Van Oss, S.B.; Swerdlow, S.H. Double-hit B-cell lymphomas with BCL6 and MYC translocations are aggressive, frequently extranodal lymphomas distinct from BCL2 double-hit B-cell lymphomas. Am. J. Surg. Pathol. 2013, 37, 323–332. [Google Scholar] [CrossRef]
- Riedell, P.A.; Smith, S.M. Double hit and double expressors in lymphoma: Definition and treatment. Cancer 2018, 124, 4622–4632. [Google Scholar] [CrossRef] [Green Version]
- Bolen, C.R.; Klanova, M.; Trneny, M.; Sehn, L.H.; He, J.; Tong, J.; Paulson, J.N.; Kim, E.; Vitolo, U.; Di Rocco, A.; et al. Prognostic impact of somatic mutations in diffuse large B-cell lymphoma and relationship to cell-of-origin: Data from the phase III GOYA study. Haematologica 2020, 105, 2298–2307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falini, B.; Fizzotti, M.; Pucciarini, A.; Bigerna, B.; Marafioti, T.; Gambacorta, M.; Pacini, R.; Alunni, C.; Natali-Tanci, L.; Ugolini, B.; et al. A monoclonal antibody (MUM1p) detects expression of the MUM1/IRF4 protein in a subset of germinal center B cells, plasma cells, and activated T cells. Blood 2000, 95, 2084–2092. [Google Scholar] [CrossRef]
- Tsuboi, K.; Iida, S.; Inagaki, H.; Kato, M.; Hayami, Y.; Hanamura, I.; Miura, K.; Harada, S.; Kikuchi, M.; Komatsu, H.; et al. MUM1/IRF4 expression as a frequent event in mature lymphoid malignancies. Leukemia 2000, 14, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Saha, A.; Robertson, E.S. Mechanisms of B-Cell Oncogenesis Induced by Epstein-Barr Virus. J. Virol. 2019, 93, e00238-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munz, C. Latency and lytic replication in Epstein-Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Ahsan, N.; Kanda, T.; Nagashima, K.; Takada, K. Epstein-Barr virus transforming protein LMP1 plays a critical role in virus production. J. Virol. 2005, 79, 4415–4424. [Google Scholar] [CrossRef] [Green Version]
- Dirmeier, U.; Neuhierl, B.; Kilger, E.; Reisbach, G.; Sandberg, M.L.; Hammerschmidt, W. Latent membrane protein 1 is critical for efficient growth transformation of human B cells by epstein-barr virus. Cancer Res. 2003, 63, 2982–2989. [Google Scholar]
- Hussain, T.; Mulherkar, R. Lymphoblastoid Cell lines: A Continuous in Vitro Source of Cells to Study Carcinogen Sensitivity and DNA Repair. Int. J. Mol. Cell. Med. 2012, 1, 75–87. [Google Scholar]
- Iwakiri, D.; Takada, K. Role of EBERs in the Pathogenesis of EBV Infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar] [CrossRef]
- Tierney, R.; Kirby, H.; Nagra, J.; Rickinson, A.; Bell, A. The Epstein-Barr virus promoter initiating B-cell transformation is activated by RFX proteins and the B-cell-specific activator protein BSAP/Pax5. J. Virol. 2000, 74, 10458–10467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes-Moreno, S.; Odqvist, L.; Diaz-Perez, J.A.; Lopez, A.B.; de Villambrosía, S.G.; Mazorra, F.; Castillo, M.E.; Lopez, M.; Pajares, R.; García, J.F.; et al. EBV-positive diffuse large B-cell lymphoma of the elderly is an aggressive post-germinal center B-cell neoplasm characterized by prominent nuclear factor-kB activation. Mod. Pathol. 2012, 25, 968–982. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Gloghini, A.; Dotti, G. EBV-Associated Lymphoproliferative Disorders: Classification and Treatment. Oncologist 2008, 13, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Xu, Z.; Lin, W.; Duan, Y.; Lu, C.; Liu, W.; Su, W.; Yan, Y.; Liu, H.; Liu, L.; et al. Comprehensive Genomic Profiling of EBV-Positive Diffuse Large B-cell Lymphoma and the Expression and Clinicopathological Correlations of Some Related Genes. Front. Oncol. 2019, 9, 683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapman, J.R.; Bouska, A.C.; Zhang, W.; Alderuccio, J.P.; Lossos, I.S.; Rimsza, L.M.; Maguire, A.; Yi, S.; Chan, W.C.; Vega, F.; et al. EBV-positive HIV-associated diffuse large B cell lymphomas are characterized by JAK/STAT (STAT3) pathway mutations and unique clinicopathologic features. Br. J. Haematol. 2021, 194, 870–878. [Google Scholar] [CrossRef] [PubMed]
- Ahye, N.; Bellizzi, A.; May, D.; Wollebo, H.S. The Role of the JC Virus in Central Nervous System Tumorigenesis. Int. J. Mol. Sci. 2020, 21, 6236. [Google Scholar] [CrossRef]
- Khalili, K.; Del Valle, L.; Otte, J.; Weaver, M.; Gordon, J. Human neurotropic polyomavirus, JCV, and its role in carcinogenesis. Oncogene 2003, 22, 5181–5191. [Google Scholar] [CrossRef] [Green Version]
- Atwood, W.J.; Amemiya, K.; Traub, R.; Harms, J.; Major, E.O. Interaction of the human polyomavirus, JCV, with human B-lymphocytes. Virology 1992, 190, 716–723. [Google Scholar] [CrossRef]
- Del Valle, L.; White, M.K.; Enam, S.; Pina Oviedo, S.; Bromer, M.Q.; Thomas, R.M.; Parkman, H.P.; Khalili, K. Detection of JC virus DNA sequences and expression of viral T antigen and agnoprotein in esophageal carcinoma. Cancer 2005, 103, 516–527. [Google Scholar] [CrossRef]
- Ripple, M.J.; Parker Struckhoff, A.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Del Valle, L. Activation of c-Myc and Cyclin D1 by JCV T-Antigen and beta-catenin in colon cancer. PLoS ONE 2014, 9, e106257. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Murai, Y.; Hong, M.; Nakanishi, Y.; Nomoto, K.; Masuda, S.; Tsuneyama, K.; Takano, Y. JC virus detection in the human tissue specimens. J. Clin. Pathol. 2007, 60, 787–793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darbinyan, A.; White, M.K.; Akan, S.; Radhakrishnan, S.; Del Valle, L.; Amini, S.; Khalili, K. Alterations of DNA damage repair pathways resulting from JCV infection. Virology 2007, 364, 73–86. [Google Scholar] [CrossRef] [Green Version]
- M’Kacher, R.; Andreoletti, L.; Flamant, S.; Milliat, F.; Girinsky, T.; Dossou, J.; Violot, D.; Assaf, E.; Clausse, B.; Koscielny, S.; et al. JC human polyomavirus is associated to chromosomal instability in peripheral blood lymphocytes of Hodgkin’s lymphoma patients and poor clinical outcome. Ann. Oncol. 2010, 21, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Caracciolo, V.; Macaluso, M.; D’Agostino, L.; Montanari, M.; Scheff, J.; Reiss, K.; Khalili, K.; Giordano, A. Cross-talk between T-Ag presence and pRb family and p53/p73 signaling in mouse and human medulloblastoma. J. Cell. Biochem. 2010, 110, 182–190. [Google Scholar] [CrossRef] [Green Version]
- Del Valle, L.; White, M.K.; Khalili, K. Potential mechanisms of the human polyomavirus JC in neural oncogenesis. J. Neuropathol. Exp. Neurol. 2008, 67, 729–740. [Google Scholar] [CrossRef]
- Del Valle, L.; Gordon, J.; Assimakopoulou, M.; Enam, S.; Geddes, J.F.; Varakis, J.N.; Katsetos, C.D.; Croul, S.; Khalili, K. Detection of JC Virus DNA sequences and expression of the viral regulatory protein T-Antigen in tumors of the Central Nervous System. Cancer Res. 2001, 61, 4287–4293. [Google Scholar]
- Grogg, K.L.; Miller, R.F.; Dogan, A. HIV infection and lymphoma. J. Clin. Pathol. 2007, 60, 1365–1372. [Google Scholar] [CrossRef] [Green Version]
- Laurence, J.; Astrin, S.M. Human immunodeficiency virus induction of malignant transformation in human B lymphocytes. Proc. Natl. Acad. Sci. USA 1991, 88, 7635–7639. [Google Scholar] [CrossRef] [Green Version]
- Tada, H.; Rappaport, J.; Lashgari, M.; Amini, S.; Wong-Staal, F.; Khalili, K. Trans-activation of the JC virus late promoter by the tat protein of type 1 human immunodeficiency virus in glial cells. Proc. Natl. Acad. Sci. USA 1990, 87, 3479–3483. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, A.; Bloch, K.C.; Li, S.; Tang, Y.W.; Palmer, M.; Tyler, K.L. Dual infections of the central nervous system with Epstein-Barr virus. J. Infect. Dis. 2005, 191, 234–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadburn, A.; Chiu, A.; Lee, J.Y.; Chen, X.; Hyjek, E.; Banham, A.H.; Noy, A.; Kaplan, L.D.; Sparano, J.A.; Bhatia, K.; et al. Immunophenotypic analysis of AIDS-related diffuse large B-cell lymphoma and clinical implications in patients from AIDS Malignancies Consortium clinical trials 010 and 034. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2009, 27, 5039–5048. [Google Scholar] [CrossRef] [Green Version]
- Falini, B.; Mason, D.Y. Proteins encoded by genes involved in chromosomal alterations in lymphoma and leukemia: Clinical value of their detection by immunocytochemistry. Blood 2002, 99, 409–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, C.; Tiemann, M.; Schrader, C.; Janssen, D.; Wolf, E.; Vierbuchen, M.; Parwaresch, R.; Ernestus, K.; Plettenberg, A.; Stoehr, A.; et al. AIDS-related B-cell lymphoma (ARL): Correlation of prognosis with differentiation profiles assessed by immunophenotyping. Blood 2005, 106, 1762–1769. [Google Scholar] [CrossRef] [Green Version]
- Larocca, L.M.; Capello, D.; Rinelli, A.; Nori, S.; Antinori, A.; Gloghini, A.; Cingolani, A.; Migliazza, A.; Saglio, G.; Cammilleri-Broet, S.; et al. The Molecular and Phenotypic Profile of Primary Central Nervous System Lymphoma Identifies Distinct Categories of the Disease and Is Consistent With Histogenetic Derivation From Germinal Center–Related B Cells. Blood 1998, 92, 1011–1019. [Google Scholar] [PubMed]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Radke, J.; Ishaque, N.; Koll, R.; Gu, Z.; Schumann, E.; Sieverling, L.; Uhrig, S.; Hübschmann, D.; Toprak, U.H.; López, C.; et al. The genomic and transcriptional landscape of primary central nervous system lymphoma. Nat. Commun. 2022, 13, 2558. [Google Scholar] [CrossRef] [PubMed]
- Jeang, K.T.; August, J.T.; Murad, F. HIV-1: Molecular Biology and Pathogenesis Viral Mechanisms, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 55. [Google Scholar]
- Hajtovic, S.; Liu, C.; Diefenbach, C.M.; Placantonakis, D.G. Epstein-Barr Virus-Positive Primary Central Nervous System Lymphoma in a 40-Year-Old Immunocompetent Patient. Cureus 2021, 13, e12754. [Google Scholar] [CrossRef] [PubMed]
- Utsuki, S.; Oka, H.; Miyajima, Y.; Kijima, C.; Yasui, Y.; Fujii, K. Epstein-Barr virus (EBV)-associated primary central nervous system lymphoma: Is incidence of EBV expression associated with median survival time? Brain Tumor Pathol. 2011, 28, 145–149. [Google Scholar] [CrossRef]
- Chapagain, M.L.; Nerurkar, V.R. Human polyomavirus JC (JCV) infection of human B lymphocytes: A possible mechanism for JCV transmigration across the blood-brain barrier. J. Infect. Dis. 2010, 202, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Lafon, M.E.; Dutronc, H.; Dubois, V.; Pellegrin, I.; Barbeau, P.; Ragnaud, J.M.; Pellegrin, J.L.; Fleury, H.J. JC virus remains latent in peripheral blood B lymphocytes but replicates actively in urine from AIDS patients. J. Infect. Dis. 1998, 177, 1502–1505. [Google Scholar] [CrossRef]
- Neel, J.V.; Major, E.O.; Awa, A.A.; Glover, T.; Burgess, A.; Traub, R.; Curfman, B.; Satoh, C. Hypothesis: “Rogue cell”-type chromosomal damage in lymphocytes is associated with infection with the JC human polyoma virus and has implications for oncopenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 2690–2695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballerini, P.; Gaidano, G.; Gong, J.Z.; Tassi, V.; Saglio, G.; Knowles, D.M.; Dalla-Favera, R. Multiple Genetic Lesions in Acquired Immunodeficiency Syndrome-Related Non-Hodgkin’s Lymphoma. Blood 1993, 81, 166–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton-Dutoit, S.J.; Rea, D.; Raphael, M.; Sandvej, K.; Delecluse, H.J.; Gisselbrecht, C.; Marelle, L.; van Krieken, H.J.; Pallesen, G. Epstein-Barr virus-latent gene expression and tumor cell phenotype in acquired immunodeficiency syndrome-related non-Hodgkin’s lymphoma. Correlation of lymphoma phenotype with three distinct patterns of viral latency. Am. J. Pathol. 1993, 143, 1072–1085. [Google Scholar]
- Kilger, E.; Kieser, A.; Baumann, M.; Hammerschmidt, W. Epstein-Barr virus-mediated B-cell proliferation is dependent upon latent membrane protein 1, which simulates an activated CD40 receptor. EMBO J. 1998, 17, 1700–1709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Egan, J.D.; Ring, B.L.; Reding, M.J.; Wells, I.C.; Shuman, R.M. Reticulum cell sarcoma and Progressive Multifocal Leukoencephalopathy following renal transplantation. Transplantation 1980, 29, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Brecher, K.; Hochberg, F.H.; Louis, D.N.; de la Monte, S.; Riskind, P. Case report of unusual leukoencephalopathy preceding primary CNS lymphoma. J. Neurol. Neurosurg. Psychiatry 1998, 65, 917–920. [Google Scholar] [CrossRef] [Green Version]
- Gallia, G.L.; Del Valle, L.; Laine, C.; Curtis, M.; Khalili, K. Concomitant Progressive Multifocal Leucoencephalopathy and primary central nervous system Lymphoma expressing JC Virus oncogenic protein, large T-Antigen. Mol. Pathol. 2001, 54, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Reddi, A.; Patel, N.; Morris, N.A. Diffuse large B cell lymphoma secondary to JC Virus in Progressive Multifocal Leukoencephalopathy. J. Neurovirol. 2019, 25, 883–886. [Google Scholar] [CrossRef]
- Del Valle, L.; Enam, S.; Lara, C.; Miklossy, J.; Khalili, K.; Gordon, J. Primary central nervous system lymphoma expressing the human neurotropic polyomavirus, JC virus, genome. J. Virol. 2004, 78, 3462–3469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monaco, M.C.; Atwood, W.J.; Gravell, M.; Tornatore, C.S.; Major, E.O. JC virus infection of hematopoietic progenitor cells, primary B lymphocytes, and tonsillar stromal cells: Implications for viral latency. J. Virol. 1996, 70, 7004–7012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, V.; Dutronc, H.; Lafon, M.E.; Poinsot, V.; Pellegrin, J.L.; Ragnaud, J.M.; Ferrer, A.M.; Fleury, H.J. Latency and reactivation of JC virus in peripheral blood of human immunodeficiency virus type 1-infected patients. J. Clin. Microbiol. 1997, 35, 2288–2292. [Google Scholar] [CrossRef] [Green Version]
- Dolei, A.; Pietropaolo, V.; Gomes, E.; Di Taranto, C.; Ziccheddu, M.; Spanu, M.A.; Lavorino, C.; Manca, M.; Degener, A.M. Polyomavirus persistence in lymphocytes: Prevalence in lymphocytes from blood donors and healthy personnel of a blood transfusion centre. J. Gen. Virol. 2000, 81, 1967–1973. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Kaminski, R.; Khalili, K.; Wollebo, H.S. Rad51 activates polyomavirus JC early transcription. PLoS ONE 2014, 9, e110122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferguson, D.O.; Sekiguchi, J.M.; Chang, S.; Frank, K.M.; Gao, Y.; DePinho, R.A.; Alt, F.W. The nonhomologous end-joining pathway of DNA repair is required for genomic stability and the suppression of translocations. Proc. Natl. Acad. Sci. USA 2000, 97, 6630–6633. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.J.; Smith, S. Sister telomeres rendered dysfunctional by persistent cohesion are fused by NHEJ. J. Cell Biol. 2009, 184, 515–526. [Google Scholar] [CrossRef] [Green Version]
- Reiss, K.; Khalili, K.; Giordano, A.; Trojanek, J. JC virus large T-antigen and IGF-I signaling system merge to affect DNA repair and genomic integrity. J. Cell Physiol. 2006, 206, 295–300. [Google Scholar] [CrossRef]
- Lazutka, J.R.; Neel, J.V.; Major, E.O.; Dedonyte, V.; Mierauskine, J.; Slapsyte, G.; Kesminiene, A. High titers of antibodies to two human polyomaviruses, JCV and BKV, correlate with increased frequency of chromosomal damage in human lymphocytes. Cancer Lett. 1996, 109, 177–183. [Google Scholar] [CrossRef]
- Johnson, E.M.; Wortman, M.J.; Dagdanova, A.V.; Lundberg, P.S.; Daniel, D.C. Polyomavirus JC in the context of immunosuppression: A series of adaptive, DNA replication-driven recombination events in the development of progressive multifocal leukoencephalopathy. Clin. Dev. Immunol. 2013, 2013, 197807. [Google Scholar] [CrossRef] [Green Version]
- Pietropaolo, V.; Videtta, M.; Fioriti, D.; Mischitelli, M.; Arancio, A.; Orsi, N.; Degener, A.M. Rearrangement patterns of JC virus noncoding control region from different biological samples. J. NeuroVirol. 2003, 9, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Wortman, M.J.; Lundberg, P.S.; Dagdanova, A.V.; Venkataraman, P.; Daniel, D.C.; Johnson, E.M. Opportunistic DNA Recombination With Epstein-Barr Virus at Sites of Control Region Rearrangements Mediating JC Virus Neurovirulence. J. Infect. Dis. 2016, 213, 1436–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, C.S.; Dezube, B.J.; Bhargava, P.; Autissier, P.; Wüthrich, C.; Miller, J.; Koralnik, I.J. Detection of JC virus DNA and proteins in the bone marrow of HIV-positive and HIV-negative patients: Implications for viral latency and neurotropic transformation. J. Infect. Dis. 2009, 199, 881–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barbier, M.T.; Del Valle, L. Co-Detection of EBV and Human Polyomavirus JCPyV in a Case of AIDS-Related Multifocal Primary Central Nervous System Diffuse Large B-Cell Lymphoma. Viruses 2023, 15, 755. https://doi.org/10.3390/v15030755
Barbier MT, Del Valle L. Co-Detection of EBV and Human Polyomavirus JCPyV in a Case of AIDS-Related Multifocal Primary Central Nervous System Diffuse Large B-Cell Lymphoma. Viruses. 2023; 15(3):755. https://doi.org/10.3390/v15030755
Chicago/Turabian StyleBarbier, Mallory T., and Luis Del Valle. 2023. "Co-Detection of EBV and Human Polyomavirus JCPyV in a Case of AIDS-Related Multifocal Primary Central Nervous System Diffuse Large B-Cell Lymphoma" Viruses 15, no. 3: 755. https://doi.org/10.3390/v15030755