
EBV Reactivation from Latency Is a Degrading Experience for the Host

Abstract
:1. Introduction
2. Host Shutoff Mechanisms Are Highly Divergent, Even among Herpesviruses
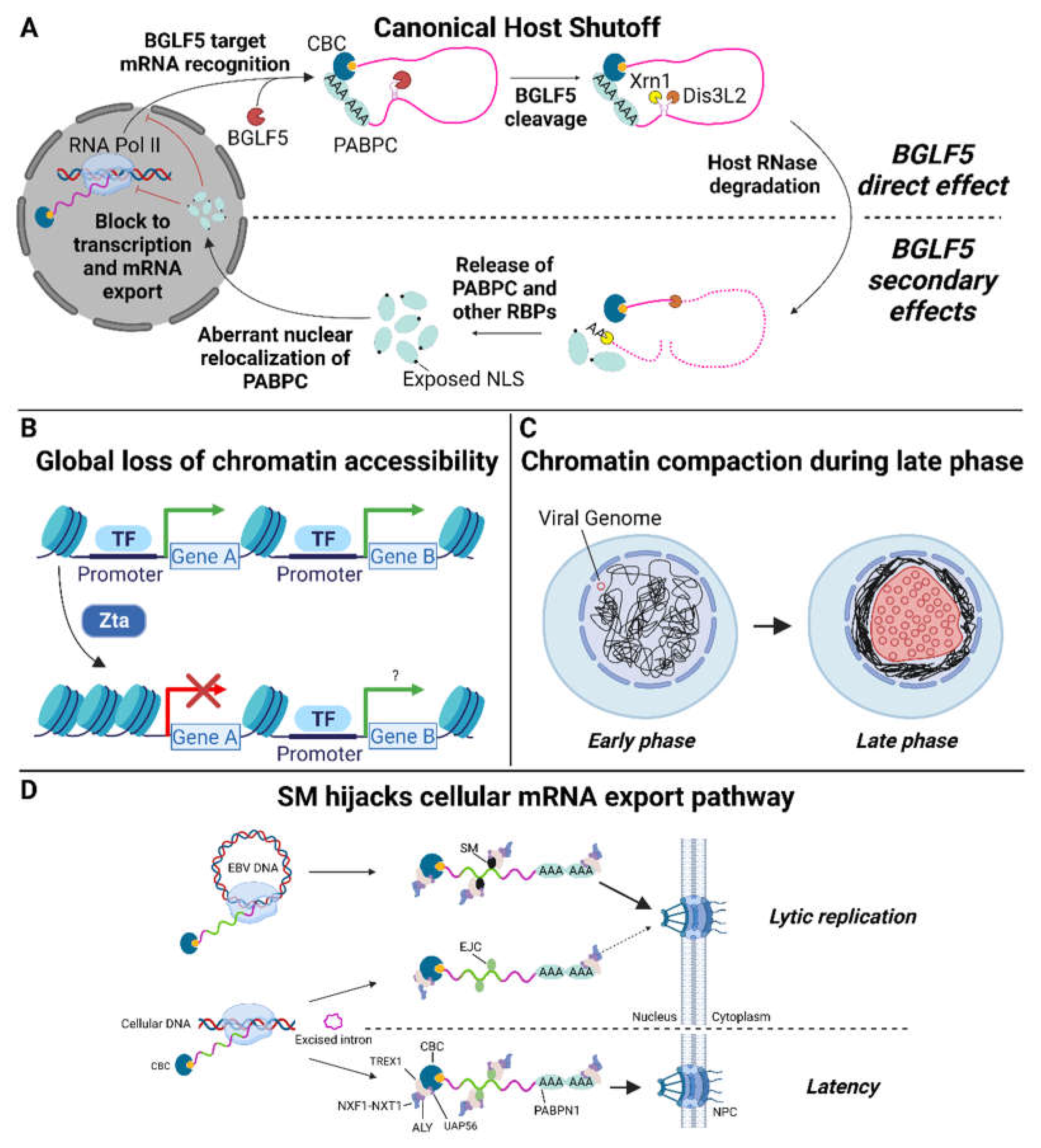
3. Host Shutoff in Epstein–Barr Virus Infection
4. Selectivity
5. Host Shutoff Escape
6. Secondary HSF Effects May Augment Shutoff
7. Host Genes Targeted by HSF during Lytic Infection
8. HSF Interactions with Viral Gene Expression
9. Non-HSF Viral Proteins Contribute to Impaired Host Gene Expression
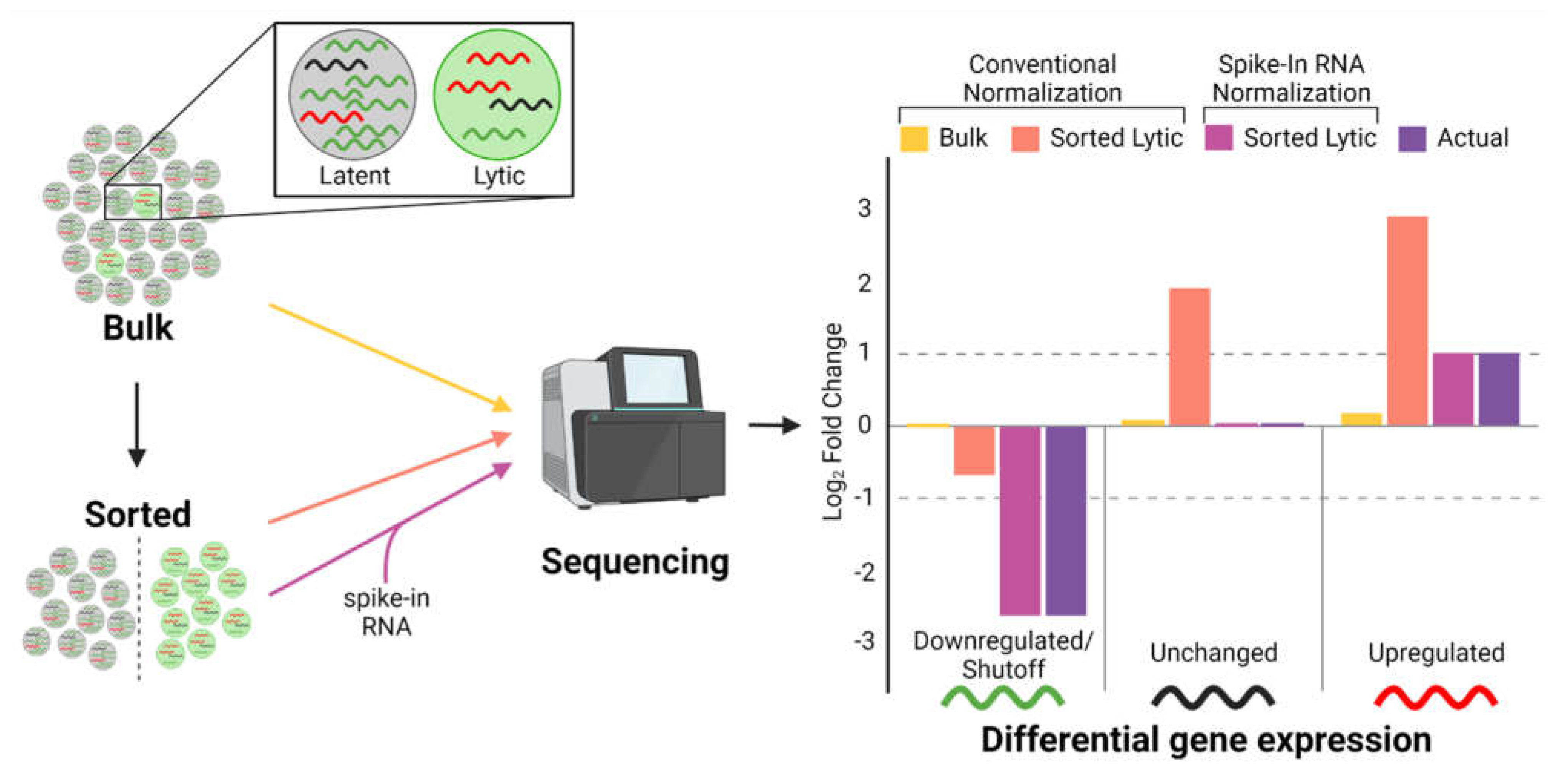
10. Technical Barriers to Defining HSF Effects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wong, Y.; Meehan, M.T.; Burrows, S.R.; Doolan, D.L.; Miles, J.J. Estimating the global burden of Epstein-Barr virus-related cancers. J. Cancer. Res. Clin. Oncol. 2022, 148, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Gewurz, B.; Longnecker, R.; Cohen, J. Epstein-Barr Virus. In Fields Virology, 7th ed.; Howley, P., Knipe, D., Cohen, J., Damania, B., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2021; pp. 324–388. [Google Scholar]
- Johannsen, E.; Kaye, K. Epstein-Barr Virus (Infectious Mononucleosis, Epstein-Barr Virus-Associated Malignant Diseases, and Other Diseases). In Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases; Bennett, J., Dolin, R., Blaser, M., Eds.; Elsevier: Philadelphia, PA, USA, 2020; pp. 1872–1890. [Google Scholar]
- Jangra, S.; Yuen, K.S.; Botelho, M.G.; Jin, D.Y. Epstein-Barr Virus and Innate Immunity: Friends or Foes? Microorganisms 2019, 7, 183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fenwick, M. The Effects of Herpesviruses on Cellular Macromolecular Synthesis. In Viral Cytopathology. Comprehensive Virology, 7th ed.; Fraenkel-Conrat, H., Wagner, R., Eds.; Plenum Press: New York, NY, USA, 1984; pp. 359–390. [Google Scholar]
- Ball, C.B.; Parida, M.; Li, M.; Spector, B.M.; Suarez, G.A.; Meier, J.L.; Price, D.H. Human Cytomegalovirus Infection Elicits Global Changes in Host Transcription by RNA Polymerases I, II, and III. Viruses 2022, 14, 779. [Google Scholar] [CrossRef]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and viral proteins in the virion of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 4952–4964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortz, E.; Whitelegge, J.P.; Jia, Q.; Zhou, Z.H.; Stewart, J.P.; Wu, T.T.; Sun, R. Identification of proteins associated with murine gammaherpesvirus 68 virions. J. Virol. 2003, 77, 13425–13432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [Green Version]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.H.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [Green Version]
- Glaunsinger, B.; Chavez, L.; Ganem, D. The exonuclease and host shutoff functions of the SOX protein of Kaposi’s sarcoma-associated herpesvirus are genetically separable. J. Virol. 2005, 79, 7396–7401. [Google Scholar] [CrossRef] [Green Version]
- Richner, J.M.; Clyde, K.; Pezda, A.C.; Cheng, B.Y.H.; Wang, T.; Kumar, G.R.; Covarrubias, S.; Coscoy, L.; Glaunsinger, B. Global mRNA degradation during lytic gammaherpesvirus infection contributes to establishment of viral latency. PLoS Pathog. 2011, 7, e1002150. [Google Scholar] [CrossRef] [Green Version]
- Zuo, J.; Thomas, W.; van Leeuwen, D.; Middeldorp, J.M.; Wiertz, E.J.H.J.; Ressing, M.E.; Rowe, M. The DNase of gammaherpesviruses impairs recognition by virus-specific CD8+ T cells through an additional host shutoff function. J. Virol. 2008, 82, 2385–2393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, S.; Richner, J.M.; Clyde, K.; Lee, Y.J.; Glaunsinger, B.A. Host shutoff is a conserved phenotype of gammaherpesvirus infection and is orchestrated exclusively from the cytoplasm. J. Virol. 2009, 83, 9554–9566. [Google Scholar] [CrossRef] [Green Version]
- Bagneris, C.; Briggs, L.C.; Savva, R.; Ebrahimi, B.; Barrett, T.E. Crystal structure of a KSHV-SOX-DNA complex: Insights into the molecular mechanisms underlying DNase activity and host shutoff. Nucleic Acids Res. 2011, 39, 5744–5756. [Google Scholar] [CrossRef] [Green Version]
- Buisson, M.; Géoui, T.; Flot, D.; Tarbouriech, N.; Ressing, M.E.; Wiertz, E.J.; Burmeister, W.P. A bridge crosses the active-site canyon of the Epstein-Barr virus nuclease with DNase and RNase activities. J. Mol. Biol. 2009, 391, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Abernathy, E.; Gilbertson, S.; Alla, R.; Glaunsinger, B. Viral Nucleases Induce an mRNA Degradation-Transcription Feedback Loop in Mammalian Cells. Cell Host Microbe 2015, 18, 243–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, S.; Gaglia, M.M.; Kumar, G.R.; Wong, W.; Jackson, A.O.; Glaunsinger, B.A. Coordinated destruction of cellular messages in translation complexes by the gammaherpesvirus host shutoff factor and the mammalian exonuclease Xrn1. PLoS Pathog. 2011, 7, e1002339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaglia, M.M.; Covarrubias, S.; Wong, W.; Glaunsinger, B.A. A common strategy for host RNA degradation by divergent viruses. J. Virol. 2012, 86, 9527–9530. [Google Scholar] [CrossRef] [Green Version]
- Abernathy, E.; Clyde, K.; Yeasmin, R.; Krug, L.; Burlingame, A.; Coscoy, L.; Glaunsinger, B. Gammaherpesviral gene expression and virion composition are broadly controlled by accelerated mRNA degradation. PLoS Pathog. 2014, 10, e1003882. [Google Scholar] [CrossRef] [Green Version]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcriptome-Wide Cleavage Site Mapping on Cellular mRNAs Reveals Features Underlying Sequence-Specific Cleavage by the Viral Ribonuclease SOX. PLoS Pathog. 2015, 11, e1005305. [Google Scholar] [CrossRef] [Green Version]
- Mendez, A.S.; Vogt, C.; Bohne, J.; Glaunsinger, B.A. Site specific target binding controls RNA cleavage efficiency by the Kaposi’s sarcoma-associated herpesvirus endonuclease SOX. Nucleic Acids Res. 2018, 46, 11968–11979. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.; Patschull, A.O.; Bagnéris, C.; Ryan, H.; Sanderson, C.M.; Ebrahimi, B.; Nobeli, I.; Barrett, T.E. KSHV SOX mediated host shutoff: The molecular mechanism underlying mRNA transcript processing. Nucleic Acids Res. 2017, 45, 4756–4767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horst, D.; Burmeister, W.P.; Boer, I.G.J.; van Leeuwen, D.; Buisson, M.; Gorbalenya, A.E.; Wiertz, E.J.H.J.; Ressing, M.E. The “Bridge” in the Epstein-Barr virus alkaline exonuclease protein BGLF5 contributes to shutoff activity during productive infection. J. Virol. 2012, 86, 9175–9187. [Google Scholar] [CrossRef] [Green Version]
- Feederle, R.; Mehl-Lautscham, A.M.; Bannert, H.; Delecluse, H.J. The Epstein-Barr virus protein kinase BGLF4 and the exonuclease BGLF5 have opposite effects on the regulation of viral protein production. J. Virol. 2009, 83, 10877–10891. [Google Scholar] [CrossRef] [Green Version]
- Glaunsinger, B.; Ganem, D. Highly selective escape from KSHV-mediated host mRNA shutoff and its implications for viral pathogenesis. J. Exp. Med. 2004, 200, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hutin, S.; Lee, Y.; Glaunsinger, B.A. An RNA element in human interleukin 6 confers escape from degradation by the gammaherpesvirus SOX protein. J. Virol. 2013, 87, 4672–4682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.; Glaunsinger, B.A. Nuclease escape elements protect messenger RNA against cleavage by multiple viral endonucleases. PLoS Pathog. 2017, 13, e1006593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, W.; Srivastav, K.; Muller, M. C19ORF66 Broadly Escapes Virus-Induced Endonuclease Cleavage and Restricts Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2019, 93, e00373-19. [Google Scholar] [CrossRef] [Green Version]
- Clyde, K.; Glaunsinger, B.A. Deep sequencing reveals direct targets of gammaherpesvirus-induced mRNA decay and suggests that multiple mechanisms govern cellular transcript escape. PLoS ONE 2011, 6, e19655. [Google Scholar] [CrossRef]
- Macveigh-Fierro, D.; Cicerchia, A.; Cadorette, A.; Sharma, V.; Muller, M. The m(6)A reader YTHDC2 is essential for escape from KSHV SOX-induced RNA decay. Proc. Natl. Acad. Sci. USA 2022, 119, e2116662119. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Hutin, S.; Marigold, O.; Li, K.H.; Burlingame, A.; Glaunsinger, B.A. A ribonucleoprotein complex protects the interleukin-6 mRNA from degradation by distinct herpesviral endonucleases. PLoS Pathog. 2015, 11, e1004899. [Google Scholar] [CrossRef] [Green Version]
- Ersing, I.; Nobre, L.; Wang, L.W.; Soday, L.; Ma, Y.; Paulo, J.A.; Narita, Y.; Ashbaugh, C.W.; Jiang, C.; Grayson, N.E.; et al. A Temporal Proteomic Map of Epstein-Barr Virus Lytic Replication in B Cells. Cell Rep. 2017, 19, 1479–1493. [Google Scholar] [CrossRef] [Green Version]
- Kumar, G.R.; Glaunsinger, B.A. Nuclear import of cytoplasmic poly(A) binding protein restricts gene expression via hyperadenylation and nuclear retention of mRNA. Mol. Cell. Biol. 2010, 30, 4996–5008. [Google Scholar] [CrossRef] [Green Version]
- Park, R.; El-Guindy, A.; Heston, L.; Lin, S.-F.; Yu, K.-P.; Nagy, M.; Borah, S.; Delecluse, H.-J.; Steitz, J.; Miller, G. Nuclear translocation and regulation of intranuclear distribution of cytoplasmic poly(A)-binding protein are distinct processes mediated by two Epstein Barr virus proteins. PLoS ONE 2014, 9, e92593. [Google Scholar] [CrossRef]
- Kumar, G.R.; Shum, L.; Glaunsinger, B.A. Importin alpha-mediated nuclear import of cytoplasmic poly(A) binding protein occurs as a direct consequence of cytoplasmic mRNA depletion. Mol. Cell. Biol. 2011, 31, 3113–3125. [Google Scholar] [CrossRef] [Green Version]
- Van Gent, M.; Gram, A.M.; Boer, I.G.; Geerdink, R.J.; Lindenbergh, M.F.; Lebbink, R.J.; Wiertz, E.J.; Ressing, M.E. Silencing the shutoff protein of Epstein-Barr virus in productively infected B cells points to (innate) targets for immune evasion. J. Gen. Virol 2015, 96 (Pt 4), 858–865. [Google Scholar] [CrossRef] [PubMed]
- van Gent, M.; Griffin, B.D.; Berkhoff, E.G.; van Leeuwen, D.; Boer, I.G.; Buisson, M.; Hartgers, F.C.; Burmeister, W.P.; Wiertz, E.J.; Ressing, M.E. EBV lytic-phase protein BGLF5 contributes to TLR9 downregulation during productive infection. J. Immunol. 2011, 186, 1694–1702. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.; Jones, C.; Williams, M.V. The EBV-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiola, S.; Gosselin, D.; Takada, K.; Gosselin, J. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol. 2010, 185, 3620–3631. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [Green Version]
- Ladell, K.; Dorner, M.; Zauner, L.; Berger, C.; Zucol, F.; Bernasconi, M.; Niggli, F.K.; Speck, R.F.; Nadal, D. Immune activation suppresses initiation of lytic Epstein-Barr virus infection. Cell. Microbiol. 2007, 9, 2055–2069. [Google Scholar] [CrossRef] [PubMed]
- Zauner, L.; Melroe, G.T.; Sigrist, J.A.; Rechsteiner, M.P.; Dorner, M.; Arnold, M.; Berger, C.; Bernasconi, M.; Schaefer, B.W.; Speck, R.F.; et al. TLR9 triggering in Burkitt’s lymphoma cell lines suppresses the EBV BZLF1 transcription via histone modification. Oncogene 2010, 29, 4588–4598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumperz, J.E. The ins and outs of CD1 molecules: Bringing lipids under immunological surveillance. Traffic 2006, 7, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, B.K.; Tsai, K.; Allan, L.L.; Zheng, D.J.; Nie, J.C.; Biggs, C.; Hasan, M.R.; Kozak, F.K.; Elzen, P.V.D.; Priatel, J.; et al. Innate immune control of EBV-infected B cells by invariant natural killer T cells. Blood 2013, 122, 2600–2608. [Google Scholar] [CrossRef] [Green Version]
- Long, H.M.; Meckiff, B.J.; Taylor, G.S. The T-cell Response to Epstein-Barr Virus-New Tricks From an Old Dog. Front. Immunol. 2019, 10, 2193. [Google Scholar] [CrossRef]
- Quinn, L.L.; Zuo, J.; Abbott, R.; Shannon-Lowe, C.; Tierney, R.J.; Hislop, A.D.; Rowe, M. Cooperation between Epstein-Barr virus immune evasion proteins spreads protection from CD8+ T cell recognition across all three phases of the lytic cycle. PLoS Pathog. 2014, 10, e1004322. [Google Scholar] [CrossRef]
- Casco, A.; Gupta, A.; Hayes, M.; Djavadian, R.; Ohashi, M.; Johannsen, E. Accurate Quantification of Overlapping Herpesvirus Transcripts from RNA Sequencing Data. J. Virol. 2022, 96, e0163521. [Google Scholar] [CrossRef] [PubMed]
- Djavadian, R.; Hayes, M.; Johannsen, E. CAGE-seq analysis of Epstein-Barr virus lytic gene transcription: 3 kinetic classes from 2 mechanisms. PLoS Pathog. 2018, 14, e1007114. [Google Scholar] [CrossRef] [Green Version]
- Feederle, R.; Bannert, H.; Lips, H.; Muller-Lantzsch, N.; Delecluse, H.J. The Epstein-Barr virus alkaline exonuclease BGLF5 serves pleiotropic functions in virus replication. J. Virol. 2009, 83, 4952–4962. [Google Scholar] [CrossRef] [Green Version]
- Traylen, C.; Ramasubramanyan, S.; Zuo, J.; Rowe, M.; Almohammad, R.; Heesom, K.; Sweet, S.M.M.; Matthews, D.A.; Sinclair, A.J. Identification of Epstein-Barr Virus Replication Proteins in Burkitt’s Lymphoma Cells. Pathogens 2015, 4, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Liao, G.; Shan, L.; Zhang, J.; Chen, M.-R.; Hayward, G.S.; Hayward, S.D.; Desai, P.; Zhu, H. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 2009, 83, 5219–5231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Guindy, A.; Lopez-Giraldez, F.; Delecluse, H.J.; McKenzie, J.; Miller, G. A locus encompassing the Epstein-Barr virus bglf4 kinase regulates expression of genes encoding viral structural proteins. PLoS Pathog. 2014, 10, e1004307. [Google Scholar] [CrossRef] [Green Version]
- Buschle, A.; Mrozek-Gorska, P.; Cernilogar, F.M.; Ettinger, A.; Pich, D.; Krebs, S.; Mocanu, B.; Blum, H.; Schotta, G.; Straub, T.; et al. Epstein-Barr virus inactivates the transcriptome and disrupts the chromatin architecture of its host cell in the first phase of lytic reactivation. Nucleic Acids Res. 2021, 49, 3217–3241. [Google Scholar] [CrossRef] [PubMed]
- Frey, T.R.; Brathwaite, J.; Li, X.; Burgula, S.; Akinyemi, I.A.; Agarwal, S.; Burton, E.M.; Ljungman, M.; McIntosh, M.T.; Bhaduri-McIntosh, S. Nascent Transcriptomics Reveal Cellular Prolytic Factors Upregulated Upstream of the Latent-to-Lytic Switch Protein of Epstein-Barr Virus. J. Virol. 2020, 94, e01966-19. [Google Scholar] [CrossRef]
- Mure, F.; Panthu, B.; Zanella-Cléon, I.; Delolme, F.; Manet, E.; Ohlmann, T.; Gruffat, H. Epstein-Barr Virus Protein EB2 Stimulates Translation Initiation of mRNAs through Direct Interactions with both Poly(A)-Binding Protein and Eukaryotic Initiation Factor 4G. J. Virol. 2018, 92, e01917-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batisse, J.; Manet, E.; Middeldorp, J.; Sergeant, A.; Gruffat, H. Epstein-Barr virus mRNA export factor EB2 is essential for intranuclear capsid assembly and production of gp350. J. Virol. 2005, 79, 14102–14111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farjot, G.; Buisson, M.; Duc Dodon, M.; Gazzolo, L.; Sergeant, A.; Mikaelian, I. Epstein-Barr virus EB2 protein exports unspliced RNA via a Crm-1-independent pathway. J. Virol. 2000, 74, 6068–6076. [Google Scholar] [CrossRef] [Green Version]
- Gruffat, H.; Batisse, J.; Pich, D.; Neuhierl, B.; Manet, E.; Hammerschmidt, W.; Sergeant, A. Epstein-Barr virus mRNA export factor EB2 is essential for production of infectious virus. J. Virol. 2002, 76, 9635–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Z.; Marendy, E.; Wang, Y.D.; Yuan, J.; Sample, J.T.; Swaminathan, S. Multiple roles of Epstein-Barr virus SM protein in lytic replication. J. Virol. 2007, 81, 4058–4069. [Google Scholar] [CrossRef] [Green Version]
- Ruvolo, V.; Gupta, A.K.; Swaminathan, S. Epstein-Barr virus SM protein interacts with mRNA in vivo and mediates a gene-specific increase in cytoplasmic mRNA. J. Virol. 2001, 75, 6033–6041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruvolo, V.; Wang, E.; Boyle, S.; Swaminathan, S. The Epstein-Barr virus nuclear protein SM is both a post-transcriptional inhibitor and activator of gene expression. Proc. Natl. Acad. Sci. USA 1998, 95, 8852–8857. [Google Scholar] [CrossRef] [Green Version]
- Chiu, Y.F.; Sugden, A.U.; Sugden, B. Epstein-Barr viral productive amplification reprograms nuclear architecture, DNA replication, and histone deposition. Cell Host Microbe 2013, 14, 607–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Kirschstein, E.; Sugden, B. How Epstein-Barr Virus Induces the Reorganization of Cellular Chromatin. mBio 2023, 14, e0268622. [Google Scholar] [CrossRef]
- Ramasubramanyan, S.; Osborn, K.; Al-Mohammad, R.; Perez-Fernandez, I.B.N.; Zuo, J.; Balan, N.; Godfrey, A.; Patel, H.; Peters, G.; Rowe, M.; et al. Epstein-Barr virus transcription factor Zta acts through distal regulatory elements to directly control cellular gene expression. Nucleic Acids Res. 2015, 43, 3563–3577. [Google Scholar] [CrossRef] [PubMed]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.W.; Keating, S.; Gomez, R.; Franken, K.L.M.C.; Ottenhoff, T.H.M.; Spriggs, M.; Schumacher, T.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA class II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.Y.; Lufkin, T. Development of the “Three-step MACS”: A novel strategy for isolating rare cell populations in the absence of known cell surface markers from complex animal tissue. J. Biomol. Tech. 2012, 23, 69–77. [Google Scholar] [CrossRef]
- Wattrus, S.J.; Zon, L.I. A Transgenic System for Rapid Magnetic Enrichment of Rare Embryonic Cells. Zebrafish 2020, 17, 354–357. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Auburn, H.; Zuckerman, M.; Smith, M. Analysis of Epstein-Barr virus and cellular gene expression during the early phases of Epstein-Barr virus lytic induction. J. Med. Microbiol. 2016, 65, 1243–1252. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Cahir-McFarland, E.; Zhao, B.; Kieff, E. Virus and cell RNAs expressed during Epstein-Barr virus replication. J. Virol. 2006, 80, 2548–2565. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| virus | VHS | AE | |
|---|---|---|---|
| alpha | HSV | UL41 | UL12 |
| VZV | ORF17 | ORF48 | |
| beta | CMV | – | UL98 |
| HHV6 | – | U70 | |
| HHV7 | – | U70 | |
| gamma | EBV | – | BGLF5 |
| KSHV | – | ORF37/SOX |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casco, A.; Johannsen, E. EBV Reactivation from Latency Is a Degrading Experience for the Host. Viruses 2023, 15, 726. https://doi.org/10.3390/v15030726
Casco A, Johannsen E. EBV Reactivation from Latency Is a Degrading Experience for the Host. Viruses. 2023; 15(3):726. https://doi.org/10.3390/v15030726
Chicago/Turabian StyleCasco, Alejandro, and Eric Johannsen. 2023. "EBV Reactivation from Latency Is a Degrading Experience for the Host" Viruses 15, no. 3: 726. https://doi.org/10.3390/v15030726