

The Viral G-Protein-Coupled Receptor Homologs M33 and US28 Promote Cardiac Dysfunction during Murine Cytomegalovirus Infection
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell and Virus Preparation
2.2. In Vivo MCMV Infection
2.3. In Vivo Acute Replication
2.4. Viral DNA Quantification
2.5. Cardiac Function
2.6. Ex Vivo MCMV Reactivation
2.7. Histology
2.8. Cellular Gene Expression
2.9. Flow Cytometry
2.10. Statistics
3. Results
3.1. Characterization of MCMV-Associated Cardiac Dysfunction
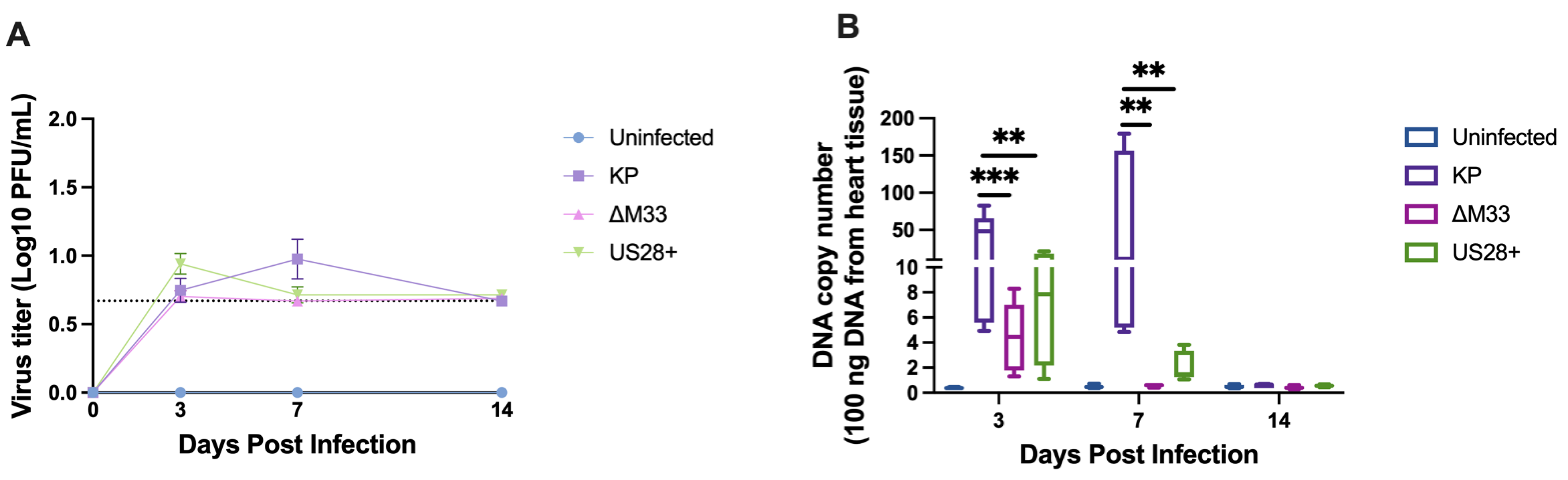
3.2. Acute Replication Kinetics and Viral DNA Load
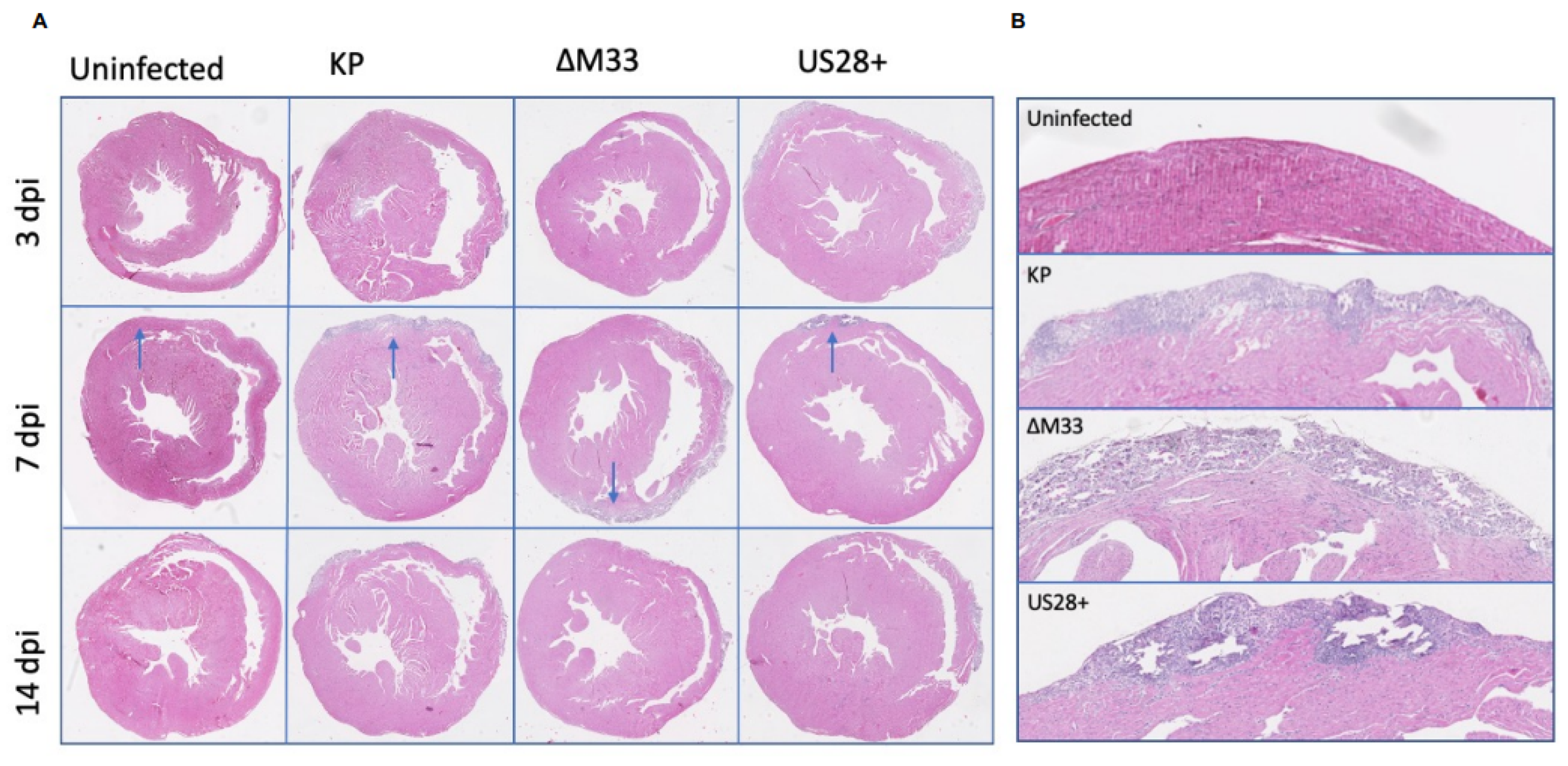
3.3. Acute Pathology
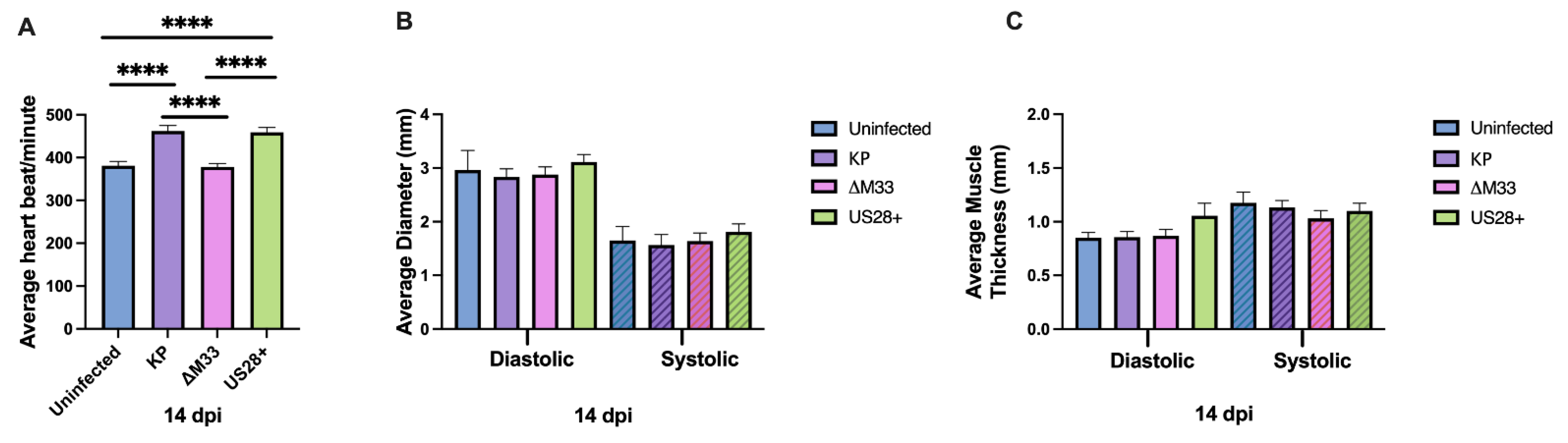
3.4. Acute Cardiac Dysfunction
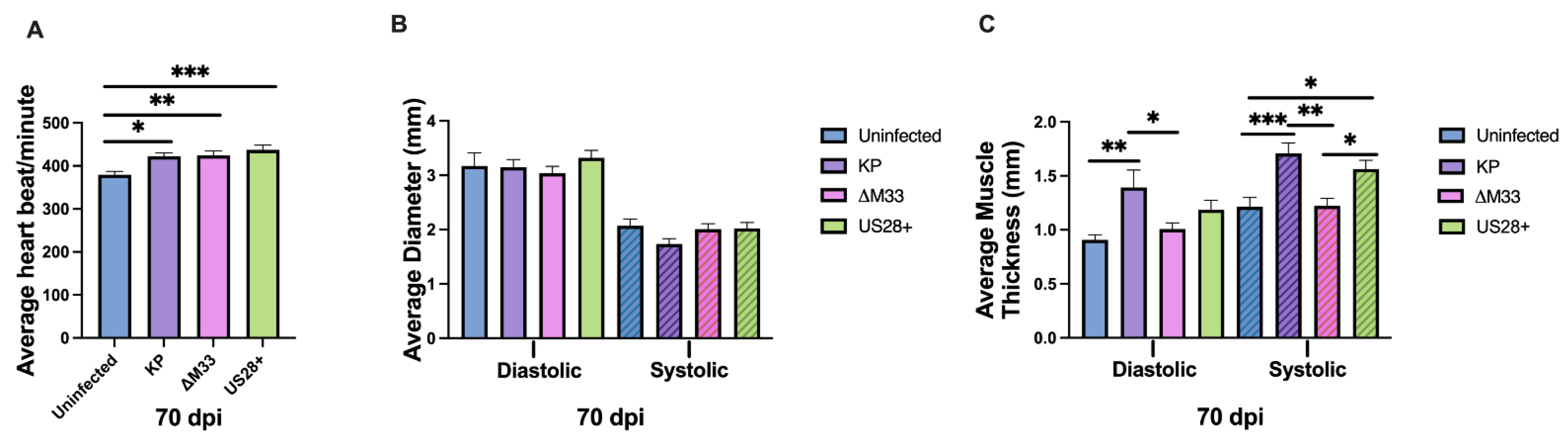
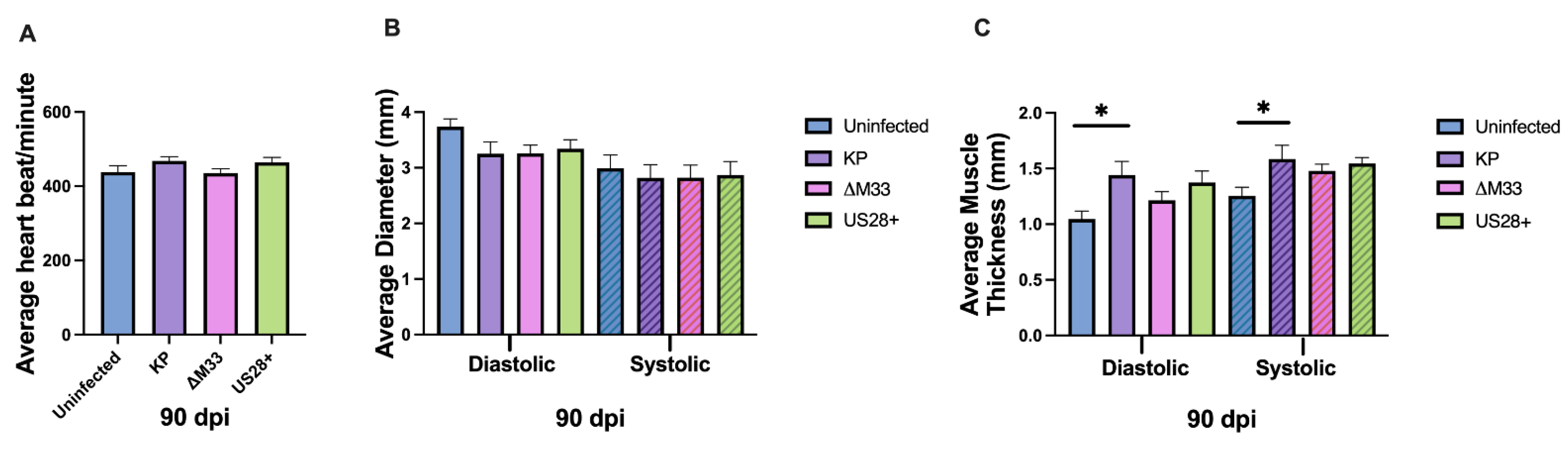
3.5. Cardiac Damage and Dysfunction during Latency
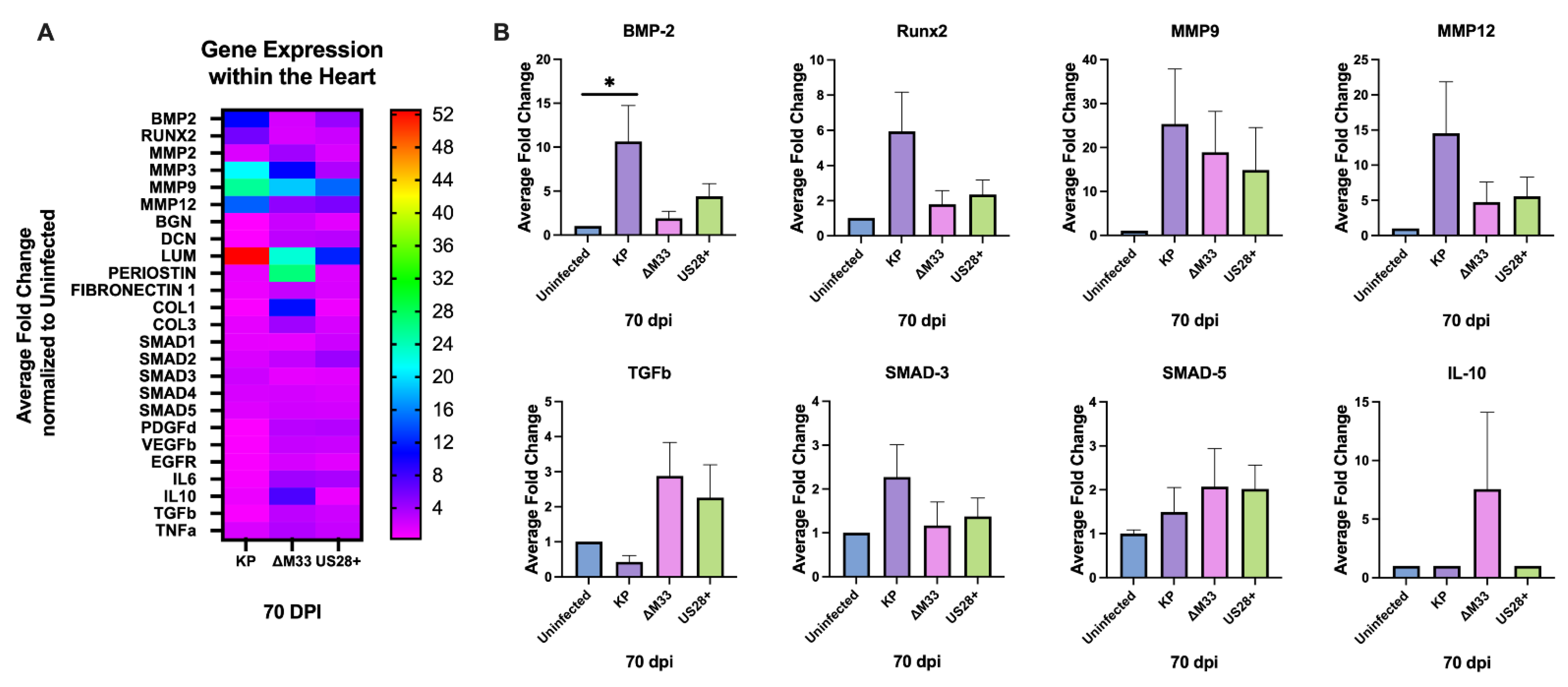
3.6. Modulation of Cardiac Gene Expression
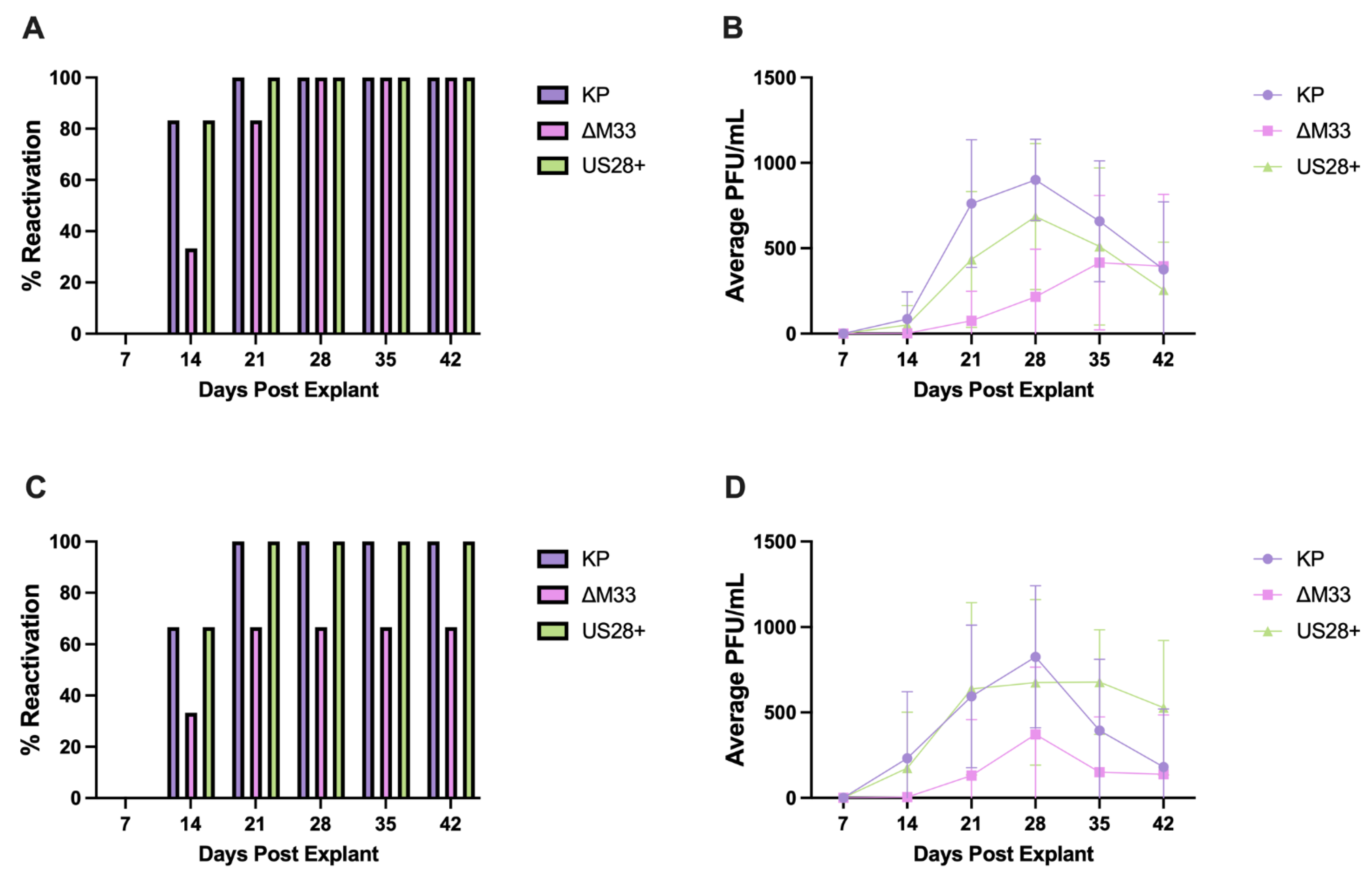
3.7. MCMV Reactivation and Immune Memory
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soderberg-Naucler, C. Does cytomegalovirus play a causative role in the development of various inflammatory diseases and cancer? J. Intern. Med. 2006, 259, 219–246. [Google Scholar] [CrossRef]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal Human Cytomegalovirus Transmission Correlates with Delayed Maternal Antibodies to gH/gL/pUL128-130-131 Complex during Primary Infection. PLoS ONE 2013, 8, e59863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padala, S.K.; Kumar, A.; Padala, S. Fulminant Cytomegalovirus Myocarditis in an Immunocompetent Host: Resolution with Oral Valganciclovir. Tex. Heart Inst. J. 2014, 41, 523–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oka, K.; Oohira, K.; Yatabe, Y.; Tanaka, T.; Kurano, K.; Kosugi, R.; Murata, M.; Hakozaki, H.; Nishikawa, T.; Tsutsumi, Y. Fulminant myocarditis demonstrating uncommon morphology?a report of two autopsy cases. Virchows Arch. 2005, 446, 259–264. [Google Scholar] [CrossRef]
- Kyto, V.; Vuorinen, T.; Saukko, P.; Lautenschlager, I.; Lignitz, E.; Saraste, A.; Voipio-Pulkki, L.-M. Cytomegalovirus Infection of the Heart Is Common in Patients with Fatal Myocarditis. Clin. Infect. Dis. 2005, 40, 683–688. [Google Scholar] [CrossRef] [PubMed]
- Blum, A.; Peleg, A.; Weinberg, M. Anti-cytomegalovirus (CMV) IgG antibody titer in patients with risk factors to atherosclerosis. Clin. Exp. Med. 2003, 3, 157–160. [Google Scholar] [CrossRef]
- Cheng, J.; Ke, Q.; Jin, Z.; Wang, H.; Kocher, O.; Morgan, J.P.; Zhang, J.; Crumpacker, C.S. Cytomegalovirus Infection Causes an Increase of Arterial Blood Pressure. PLoS Pathog. 2009, 5, e1000427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, I.; Andersson, R.; Friman, V.; Selimovic, N.; Hanzen, L.; Nasic, S.; Nyström, U.; Sigurdardottir, V. Cytomegalovirus infection and disease reduce 10-year cardiac allograft vasculopathy-free survival in heart transplant recipients. BMC Infect. Dis. 2015, 15, 582. [Google Scholar] [CrossRef] [Green Version]
- Koskinen, P.K.; Kallio, E.A.; Tikkanen, J.M.; Sihvola, R.K.; Hayry, P.J.; Lemstrom, K.B. Cytomegalovirus infection and cardiac allograft vasculopathy. Transpl. Infect. Dis. 1999, 1, 115–126. [Google Scholar] [CrossRef]
- Magno Palmeira, M.; Umemura Ribeiro, H.Y.; Garcia Lira, Y.; Machado Jucá Neto, F.O.; da Silva Rodrigues, I.A.; Fernandes da Paz, L.N.; Nascimento Pinheiro, M.D.C. Heart failure due to cytomegalovirus myocarditis in immunocompetent young adults: A case report. BMC Res. Notes 2016, 9, 391. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.H.; Bickerstaff, A.A.; Wang, J.-J.; Zimmerman, P.D.; Forster, M.R.; Nadasdy, T.; Colvin, R.B.; Hadley, G.A.; Orosz, C.G. Disruption of Murine Cardiac Allograft Acceptance by Latent Cytomegalovirus: Disruption of Murine Cardiac Allograft. Am. J. Transplant. 2008, 9, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritter, J.T.; Tang-Feldman, Y.J.; Lochhead, G.R.; Estrada, M.; Lochhead, S.; Yu, C.; Ashton-Sager, A.; Tuteja, D.; Leutenegger, C.; Pomeroy, C. In vivo characterization of cytokine profiles and viral load during murine cytomegalovirus-induced acute myocarditis. Cardiovasc. Pathol. 2010, 19, 83–93. [Google Scholar] [CrossRef]
- Lenzo, J.C.; Mansfield, J.P.; Sivamoorthy, S.; Cull, V.S.; James, C.M. Cytokine expression in murine cytomegalovirus-induced myocarditis: Modulation with interferon-α therapy. Cell. Immunol. 2003, 223, 77–86. [Google Scholar] [CrossRef]
- Vliegen, I.; Stassen, F.; Grauls, G.; Blok, R.; Bruggeman, C. MCMV infection increases early T-lymphocyte influx in atherosclerotic lesions in apoE knockout mice. J. Clin. Virol. 2002, 25, 159–171. [Google Scholar] [CrossRef]
- Bonavita, C.M.; White, T.M.; Francis, J.; Cardin, R.D. Heart Dysfunction Following Long-Term Murine Cytomegalovirus Infection: Fibrosis, Hypertrophy, and Tachycardia. Viral Immunol. 2020, 33, 237–245. [Google Scholar] [CrossRef] [Green Version]
- Fields, B.N.; Knipe, D.M.; Howley, P.M.; Griffin, D.E. Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001. [Google Scholar]
- Lagenaur, L.A.; Manning, W.C.; Martens, C.L.; MOCARSKIl, E.S. Structure and Function of the Murine Cytomegalovirus sggl Gene: A Determinant of Viral Growth in Salivary Gland Acinar Cells. J. Virol. 1994, 68, 7717–7727. [Google Scholar] [CrossRef] [Green Version]
- Rawlinson, W.D.; Farrell, H.E.; Barrell, B.G. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 1996, 70, 8833–8849. [Google Scholar] [CrossRef] [Green Version]
- Scarborough, J.A.; Paul, J.R.; Spencer, J.V. Evolution of the ability to modulate host chemokine networks via gene duplication in human cytomegalovirus (HCMV). Infect. Genet. Evol. 2017, 51, 46–53. [Google Scholar] [CrossRef]
- Bodaghi, B.; Jones, T.R.; Zipeto, D.; Vita, C.; Sun, L.; Laurent, L.; Arenzana-Seisdedos, F.; Virelizier, J.-L.; Michelson, S. Chemokine Sequestration by Viral Chemoreceptors as a Novel Viral Escape Strategy: Withdrawal of Chemokines from the Environment of Cytomegalovirus-infected Cells. J. Exp. Med. 1998, 188, 855–866. [Google Scholar] [CrossRef] [Green Version]
- Farrell, H.E.; Abraham, A.M.; Cardin, R.D.; Sparre-Ulrich, A.H.; Rosenkilde, M.M.; Spiess, K.; Jensen, T.H.; Kledal, T.N.; Davis-Poynter, N. Partial Functional Complementation between Human and Mouse Cytomegalovirus Chemokine Receptor Homologues. J. Virol. 2011, 85, 6091–6095. [Google Scholar] [CrossRef] [Green Version]
- Frank, T.; Niemann, I.; Reichel, A.; Stamminger, T. Emerging roles of cytomegalovirus-encoded G protein-coupled receptors during lytic and latent infection. Med. Microbiol. Immunol. (Berl.) 2019, 208, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Streblow, D.N.; Orloff, S.L.; Nelson, J. The HCMV Chemokine Receptor US28 is a Potential Target in Vascular Disease. Cureent Drug Targets-Infect. Disord. 2001, 1, 151–158. [Google Scholar] [CrossRef]
- Melnychuk, R.M.; Streblow, D.N.; Smith, P.P.; Hirsch, A.J.; Pancheva, D.; Nelson, J.A. Human Cytomegalovirus-Encoded G Protein-Coupled Receptor US28 Mediates Smooth Muscle Cell Migration through Gα12. J. Virol. 2004, 78, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroceanu, L.; Matlaf, L.; Bezrookove, V.; Harkins, L.; Martinez, R.; Greene, M.; Soteropoulos, P.; Cobbs, C.S. Human Cytomegalovirus US28 Found in Glioblastoma Promotes an Invasive and Angiogenic Phenotype. Cancer Res. 2011, 71, 6643–6653. [Google Scholar] [CrossRef] [Green Version]
- Fritz, N.M.; Stamminger, T.; Ramsperger-Gleixner, M.; Kuckhahn, A.V.; Müller, R.; Weyand, M.; Heim, C. Cytomegalovirus chemokine receptor M33 knockout reduces chronic allograft rejection in a murine aortic transplant model. Transpl. Immunol. 2021, 64, 101359. [Google Scholar] [CrossRef]
- Case, R.; Sharp, E.; Benned-Jensen, T.; Rosenkilde, M.M.; Davis-Poynter, N.; Farrell, H.E. Functional Analysis of the Murine Cytomegalovirus Chemokine Receptor Homologue M33: Ablation of Constitutive Signaling Is Associated with an Attenuated Phenotype In Vivo. J. Virol. 2008, 82, 1884–1898. [Google Scholar] [CrossRef] [Green Version]
- Waldhoer, M.; Kledal, T.N.; Farrell, H.; Schwartz, T.W. Murine Cytomegalovirus (CMV) M33 and Human CMV US28 Receptors Exhibit Similar Constitutive Signaling Activities. J. Virol. 2002, 76, 8161–8168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streblow, D.N.; Soderberg-Naucler, C.; Vieira, J.; Smith, P.; Wakabayashi, E.; Ruchti, F.; Mattison, K.; Altschuler, Y.; Nelson, J.A. The Human Cytomegalovirus Chemokine Receptor US28 Mediates Vascular Smooth Muscle Cell Migration. Cell 1999, 99, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Streblow, D.N.; Vomaske, J.; Smith, P.; Melnychuk, R.; Hall, L.; Pancheva, D.; Smit, M.; Casarosa, P.; Schlaepfer, D.D.; Nelson, J.A. Human Cytomegalovirus Chemokine Receptor US28-induced Smooth Muscle Cell Migration Is Mediated by Focal Adhesion Kinase and Src. J. Biol. Chem. 2003, 278, 50456–50465. [Google Scholar] [CrossRef] [Green Version]
- Melnychuk, R.M.; Smith, P.; Kreklywich, C.N.; Ruchti, F.; Vomaske, J.; Hall, L.; Loh, L.; Nelson, J.A.; Orloff, S.L.; Streblow, D.N. Mouse Cytomegalovirus M33 Is Necessary and Sufficient in Virus-Induced Vascular Smooth Muscle Cell Migration. J. Virol. 2005, 79, 10788–10795. [Google Scholar] [CrossRef] [Green Version]
- Lollinga, W.T.; de Wit, R.H.; Rahbar, A.; Vasse, G.F.; Davoudi, B.; Diepstra, A.; Riezebos-Brilman, A.; Harmsen, M.C.; Hillebrands, J.-L.; Söderberg-Naucler, C.; et al. Human Cytomegalovirus-Encoded Receptor US28 Is Expressed in Renal Allografts and Facilitates Viral Spreading In Vitro. Transplantation 2017, 101, 531–540. [Google Scholar] [CrossRef] [PubMed]
- Beisser, P.S.; Laurent, L.; Virelizier, J.-L.; Michelson, S. Human Cytomegalovirus Chemokine Receptor Gene US28 Is Transcribed in Latently Infected THP-1 Monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardin, R.D.; Schaefer, G.C.; Allen, J.R.; Davis-Poynter, N.J.; Farrell, H.E. The M33 Chemokine Receptor Homolog of Murine Cytomegalovirus Exhibits a Differential Tissue-Specific Role during In Vivo Replication and Latency. J. Virol. 2009, 83, 7590–7601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis-Poynter, N.J.; Lynch, D.M.; Vally, H.; Shellam, G.R.; Rawlinson, W.D.; Barrell, B.G.; Farrell, H.E. Identification and characterization of a G protein-coupled receptor homolog encoded by murine cytomegalovirus. J. Virol. 1997, 71, 1521–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardin, R.D.; Bravo, F.J.; Sewell, A.P.; Cummins, J.; Flamand, L.; Juteau, J.-M.; Bernstein, D.I.; Vaillant, A. Amphipathic DNA polymers exhibit antiviral activity against systemic Murine Cytomegalovirus infection. Virol. J. 2009, 6, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almanan, M.; Raynor, J.; Sholl, A.; Wang, M.; Chougnet, C.; Cardin, R.D.; Hildeman, D.A. Tissue-specific control of latent CMV reactivation by regulatory T cells. PLoS Pathog. 2017, 13, e1006507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snyder, C.M.; Allan, J.E.; Bonnett, E.L.; Doom, C.M.; Hill, A.B. Cross-Presentation of a Spread-Defective MCMV Is Sufficient to Prime the Majority of Virus-Specific CD8+ T Cells. PLoS ONE 2010, 5, e9681. [Google Scholar] [CrossRef]
- Bonavita, C.M.; White, T.M.; Stanfield, B.A.; Cardin, R.D. Characterization of murine cytomegalovirus infection and induction of calcification in Murine Aortic Vascular Smooth Muscle Cells (MOVAS). J. Virol. Methods 2021, 297, 114270. [Google Scholar] [CrossRef]
- Li, C.; Wen, A.; Shen, B.; Lu, J.; Huang, Y.; Chang, Y. FastCloning: A highly simplified, purification-free, sequence- and ligation-independent PCR cloning method. BMC Biotechnol. 2011, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Dange, R.B.; Agarwal, D.; Masson, G.S.; Vila, J.; Wilson, B.; Nair, A.; Francis, J. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc. Res. 2014, 103, 17–27. [Google Scholar] [CrossRef]
- Lenzo, J.C.; Fairweather, D.; Cull, V.; Shellam, G.R.; James(Lawson), C.M. Characterisation of Murine Cytomegalovirus Myocarditis: Cellular Infiltration of the Heart and Virus Persistence. J. Mol. Cell. Cardiol. 2002, 34, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Maussang, D.; Langemeijer, E.; Fitzsimons, C.P.; Stigter-van Walsum, M.; Dijkman, R.; Borg, M.K.; Slinger, E.; Schreiber, A.; Michel, D.; Tensen, C.P.; et al. The Human Cytomegalovirus-Encoded Chemokine Receptor US28 Promotes Angiogenesis and Tumor Formation via Cyclooxygenase-2. Cancer Res. 2009, 69, 2861–2869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Z.-Z.; Xu, J.-G.; Zhou, Y.-H.; Zheng, J.-H.; Lin, K.-Z.; Zheng, S.-Z.; Ye, M.-S.; He, Y.; Liu, C.-B.; Xue, Z.-X. Human cytomegalovirus-encoded US28 may act as a tumor promoter in colorectal cancer. World J. Gastroenterol. 2016, 22, 2789. [Google Scholar] [CrossRef] [PubMed]
- Glass, A.M.; Coombs, W.; Taffet, S.M. Spontaneous Cardiac Calcinosis in BALB/cByJ Mice. Comp. Med. 2013, 63, 9. [Google Scholar]
- Juranic Lisnic, V.; Babic Cac, M.; Lisnic, B.; Trsan, T.; Mefferd, A.; Das Mukhopadhyay, C.; Cook, C.H.; Jonjic, S.; Trgovcich, J. Dual Analysis of the Murine Cytomegalovirus and Host Cell Transcriptomes Reveal New Aspects of the Virus-Host Cell Interface. PLoS Pathog. 2013, 9, e1003611. [Google Scholar] [CrossRef]
- Hertel, L.; Mocarski, E.S. Global Analysis of Host Cell Gene Expression Late during Cytomegalovirus Infection Reveals Extensive Dysregulation of Cell Cycle Gene Expression and Induction of Pseudomitosis Independent of US28 Function. J. Virol. 2004, 78, 11988–12011. [Google Scholar] [CrossRef] [Green Version]
- Song, R.; Fullerton, D.A.; Ao, L.; Zheng, D.; Zhao, K.; Meng, X. BMP-2 and TGF-β1 mediate biglycan-induced pro-osteogenic reprogramming in aortic valve interstitial cells. J. Mol. Med. 2015, 93, 403–412. [Google Scholar] [CrossRef] [Green Version]
- Rong, S.; Zhao, X.; Jin, X.; Zhang, Z.; Chen, L.; Zhu, Y.; Yuan, W. Vascular Calcification in Chronic Kidney Disease is Induced by Bone Morphogenetic Protein-2 via a Mechanism Involving the Wnt/β-Catenin Pathway. Cell. Physiol. Biochem. 2014, 34, 2049–2060. [Google Scholar] [CrossRef]
- Ngai, D.; Lino, M.; Bendeck, M.P. Cell-Matrix Interactions and Matricrine Signaling in the Pathogenesis of Vascular Calcification. Front. Cardiovasc. Med. 2018, 5, 174. [Google Scholar] [CrossRef] [Green Version]
- Deng, X.; Meng, X.; Li, F.; Venardos, N.; Fullerton, D.; Jaggers, J. MMP-12–Induced Pro-osteogenic Responses in Human Aortic Valve Interstitial Cells. J. Surg. Res. 2019, 235, 44–51. [Google Scholar] [CrossRef]
- Gough, P.J. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J. Clin. Investig. 2005, 116, 59–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luttun, A.; Lutgens, E.; Manderveld, A.; Maris, K.; Collen, D.; Carmeliet, P.; Moons, L. Loss of Matrix Metalloproteinase-9 or Matrix Metalloproteinase-12 Protects Apolipoprotein E–Deficient Mice Against Atherosclerotic Media Destruction but Differentially Affects Plaque Growth. Circulation 2004, 109, 1408–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.J.; Venturi, V.; Quigley, M.F.; Turula, H.; Gostick, E.; Ladell, K.; Hill, B.J.; Himelfarb, D.; Quinn, K.M.; Greenaway, H.Y.; et al. Stochastic Expansions Maintain the Clonal Stability of CD8+ T Cell Populations Undergoing Memory Inflation Driven by Murine Cytomegalovirus. J. Immunol. 2020, 204, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.; Brien, J.D.; Nikolich-Žugich, J. Inflation and Long-Term Maintenance of CD8 T Cells Responding to a Latent Herpesvirus Depend upon Establishment of Latency and Presence of Viral Antigens. J. Immunol. 2009, 183, 8077–8087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klenerman, P. The (gradual) rise of memory inflation. Immunol. Rev. 2018, 283, 99–112. [Google Scholar] [CrossRef]
- White, T.M.; Bonavita, C.M.; Stanfield, B.A.; Farrell, H.E.; Davis-Poynter, N.J.; Cardin, R.D. The CMV-encoded G protein-coupled receptors M33 and US28 play pleiotropic roles in immune evasion and alter host T cell responses. Front. Immunol. 2022, 13, 1047299. [Google Scholar] [CrossRef]
- Miles, T.F.; Spiess, K.; Jude, K.M.; Tsutsumi, N.; Burg, J.S.; Ingram, J.R.; Waghray, D.; Hjorto, G.M.; Larsen, O.; Ploegh, H.L.; et al. Viral GPCR US28 can signal in response to chemokine agonists of nearly unlimited structural degeneracy. eLife 2018, 7, e35850. [Google Scholar] [CrossRef]
- Sherrill, J.D.; Stropes, M.P.; Schneider, O.D.; Koch, D.E.; Bittencourt, F.M.; Miller, J.L.C.; Miller, W.E. Activation of Intracellular Signaling Pathways by the Murine Cytomegalovirus G Protein-Coupled Receptor M33 Occurs via PLC-β/PKC-Dependent and -Independent Mechanisms. J. Virol. 2009, 83, 8141–8152. [Google Scholar] [CrossRef] [Green Version]
- Reinhardt, B. Human cytomegalovirus-induced reduction of extracellular matrix proteins in vascular smooth muscle cell cultures: A pathomechanism in vasculopathies? J. Gen. Virol. 2006, 87, 2849–2858. [Google Scholar] [CrossRef]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E.; Smit, M.J.; Wills, M.R.; Sinclair, J.H. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Liang, J.; Tang, H.; Fang, X.; Wang, F.; Ding, Y.; Huang, H.; Zhang, H. Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int. J. Biochem. Cell Biol. 2020, 118, 105645. [Google Scholar] [CrossRef]
- Wight, T.N. A role for proteoglycans in vascular disease. Matrix Biol. 2018, 71–72, 396–420. [Google Scholar] [CrossRef]
- Perrotta, I.; Sciangula, A. Matrix Metalloproteinase-9 Expression in Calcified Human Aortic Valves: A Histopathologic, Immunohistochemical, and Ultrastructural Study. Appl. Immunohistochem. Mol. Morphol. 2016, 24, 10. [Google Scholar] [CrossRef]
- Prochnau, D.; Lehmann, M.; Straube, E.; Figulla, H.; Rödel, J. Human cytomegalovirus induces MMP-1 and MMP-3 expression in aortic smooth muscle cells. Acta Microbiol. Immunol. Hung. 2011, 58, 303–317. [Google Scholar] [CrossRef]
- Straat, K.; de Klark, R.; Gredmark-Russ, S.; Eriksson, P.; Soderberg-Naucler, C. Infection with Human Cytomegalovirus Alters the MMP-9/TIMP-1 Balance in Human Macrophages. J. Virol. 2009, 83, 830–835. [Google Scholar] [CrossRef] [Green Version]
- Cousins, S.W.; Espinosa-Heidmann, D.G.; Miller, D.M.; Pereira-Simon, S.; Hernandez, E.P.; Chien, H.; Meier-Jewett, C.; Dix, R.D. Macrophage Activation Associated with Chronic Murine Cytomegalovirus Infection Results in More Severe Experimental Choroidal Neovascularization. PLoS Pathog. 2012, 8, e1002671. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Gao, J.; Wang, G.; Fei, G. Latent cytomegalovirus infection exacerbates experimental pulmonary fibrosis by activating TGF-β1. Mol. Med. Rep. 2016, 14, 1297–1301. [Google Scholar] [CrossRef] [Green Version]
- Kania, G.; Blyszczuk, P.; Eriksson, U. Mechanisms of Cardiac Fibrosis in Inflammatory Heart Disease. Trends Cardiovasc. Med. 2009, 19, 247–252. [Google Scholar] [CrossRef]
- Suthahar, N.; Meijers, W.C.; Silljé, H.H.W.; de Boer, R.A. From Inflammation to Fibrosis—Molecular and Cellular Mechanisms of Myocardial Tissue Remodelling and Perspectives on Differential Treatment Opportunities. Curr. Heart Fail. Rep. 2017, 14, 235–250. [Google Scholar] [CrossRef] [Green Version]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonavita, C.M.; White, T.M.; Francis, J.; Farrell, H.E.; Davis-Poynter, N.J.; Cardin, R.D. The Viral G-Protein-Coupled Receptor Homologs M33 and US28 Promote Cardiac Dysfunction during Murine Cytomegalovirus Infection. Viruses 2023, 15, 711. https://doi.org/10.3390/v15030711
Bonavita CM, White TM, Francis J, Farrell HE, Davis-Poynter NJ, Cardin RD. The Viral G-Protein-Coupled Receptor Homologs M33 and US28 Promote Cardiac Dysfunction during Murine Cytomegalovirus Infection. Viruses. 2023; 15(3):711. https://doi.org/10.3390/v15030711
Chicago/Turabian StyleBonavita, Cassandra M., Timothy M. White, Joseph Francis, Helen E. Farrell, Nicholas J. Davis-Poynter, and Rhonda D. Cardin. 2023. "The Viral G-Protein-Coupled Receptor Homologs M33 and US28 Promote Cardiac Dysfunction during Murine Cytomegalovirus Infection" Viruses 15, no. 3: 711. https://doi.org/10.3390/v15030711