
Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses
 ,
,  , and
, and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Viruses and Viral Infection
2.3. Inhibitors
2.4. Drug Treatment and Plaque Assay
2.5. Cell Viability Assay
2.6. RNA Extraction and qRT-PCR Assay
2.7. Negative Strand RT-qPCR
2.8. Luciferase and Bradford Protein Assay
2.9. Proinflammatory Cytokine Quantification Assay
2.10. Statistics
3. Results
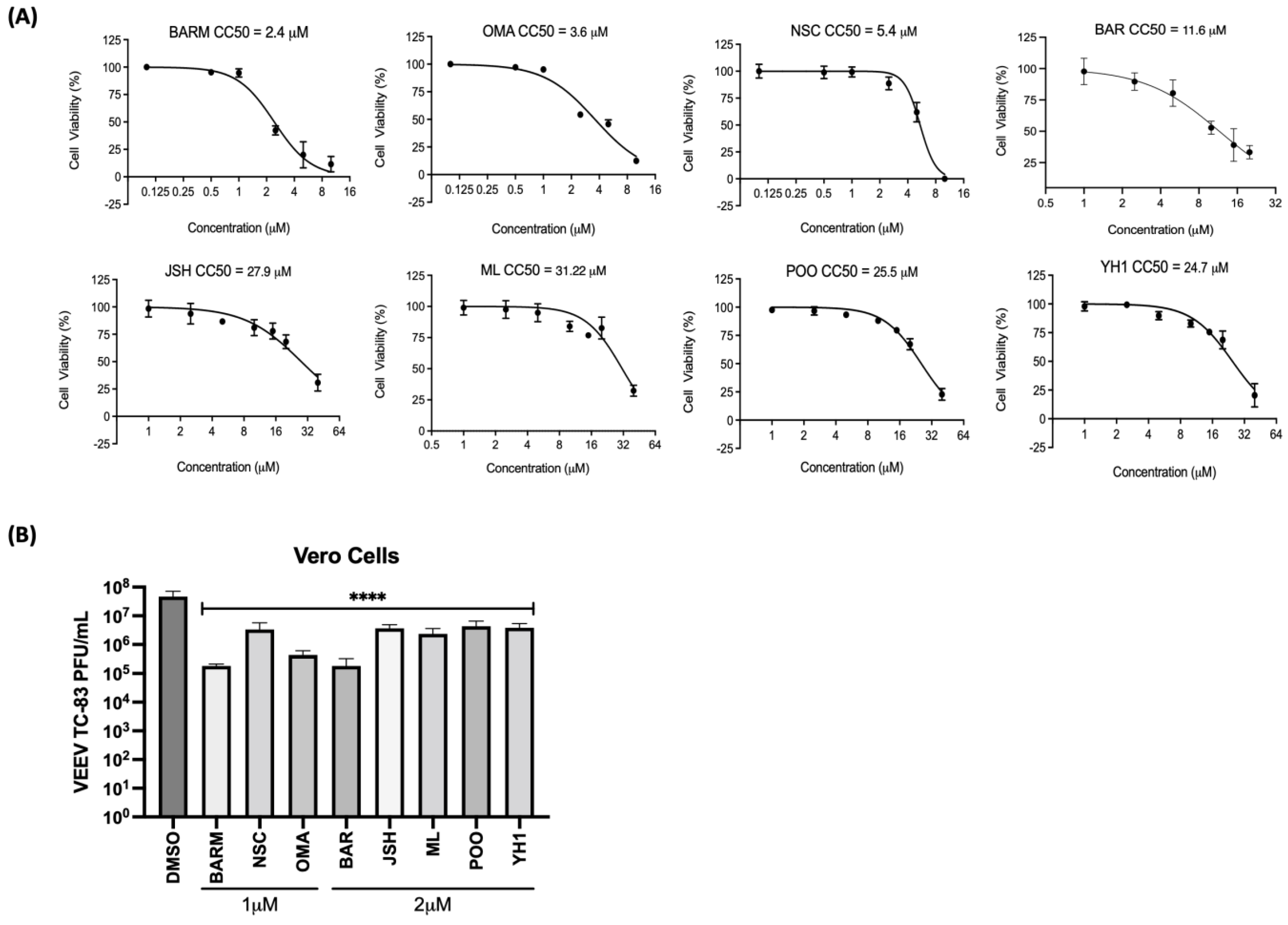
3.1. Inhibitors of UPS-Mediated Signaling Events Decrease VEEV-TC83 Load in Vero Cells
3.2. UPS-Mediated Signaling Inhibitors Demonstrate Inhibition of VEEV-TC83 in Human Astroglial (SVG-p12) and Microglial (HMC3) Cells
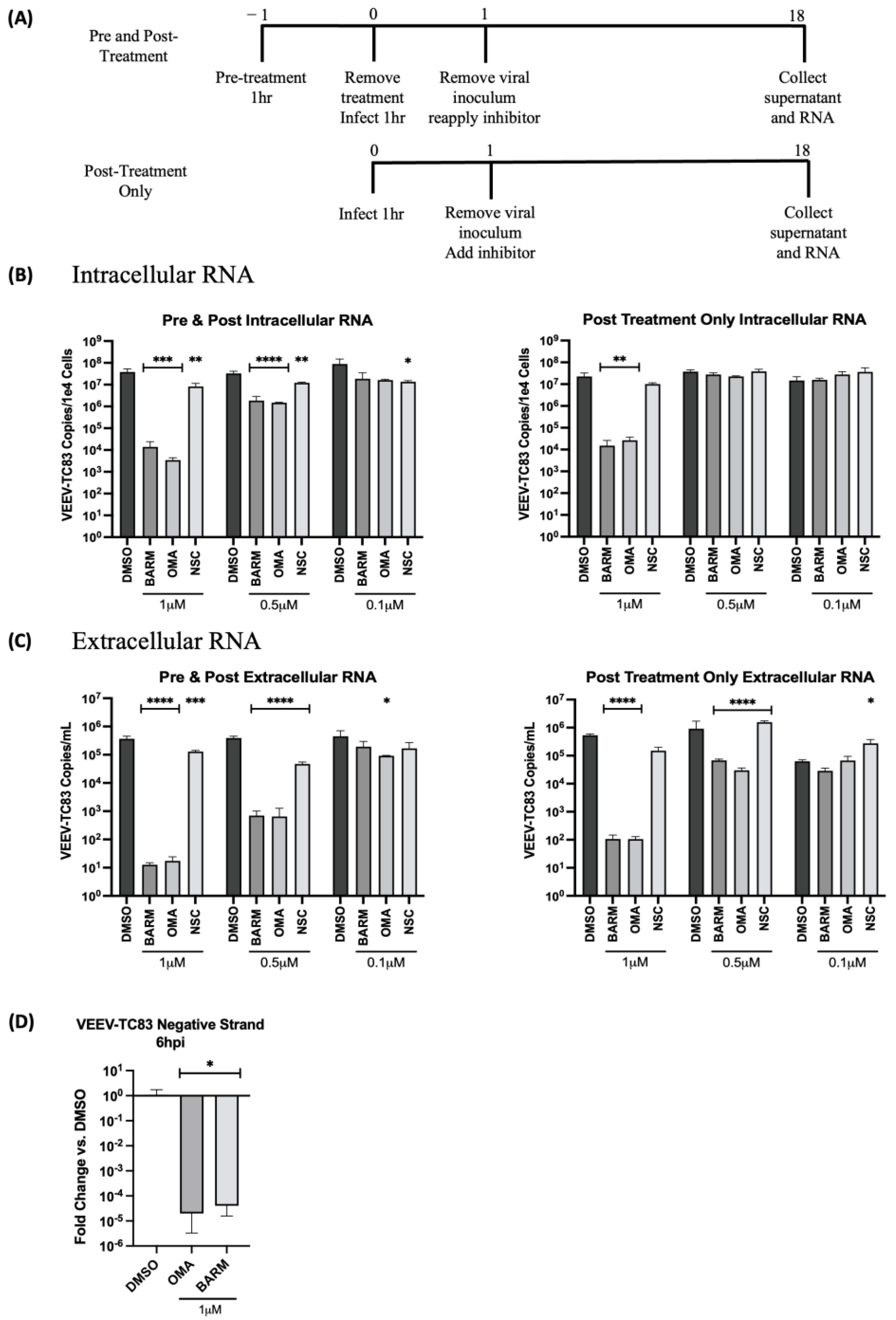
3.3. UPS Signaling Inhibitors Decrease VEEV-TC83 in a Dose-Dependent Manner
3.4. OMA, BARM and NSC Exhibit Differential Inhibitory Impact on Proinflammatory Cytokines in VEEV-TC83-Infected HMC3 Cells
3.5. OMA, BARM and NSC Exert Broad-Spectrum Viral Inhibitory Activity against Virulent Strains of VEEV and EEEV in HMC3 Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Azar, S.R.; Campos, R.K.; Bergren, N.A.; Camargos, V.N.; Rossi, S.L. Epidemic Alphaviruses: Ecology, Emergence and Outbreaks. Microorganisms 2020, 8, 1167. [Google Scholar] [CrossRef] [PubMed]
- Zacks, M.A.; Paessler, S. Encephalitic alphaviruses. Vet. Microbiol. 2010, 140, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Forrester, N.L.; Wertheim, J.O.; Dugan, V.G.; Auguste, A.J.; Lin, D.; Adams, A.P.; Chen, R.; Gorchakov, R.; Leal, G.; Estrada-Franco, J.G.; et al. Evolution and spread of Venezuelan equine encephalitis complex alphavirus in the Americas. PLoS Negl. Trop. Dis. 2017, 11, e0005693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzmán-Terán, C.; Calderón-Rangel, A.; Rodriguez-Morales, A.; Mattar, S. Venezuelan equine encephalitis virus: The problem is not over for tropical America. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 19. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Arguero, I.; Tellez-Freitas, C.M.; Weber, K.S.; Berges, B.K.; Robison, R.A.; Pickett, B.E. Alphaviruses: Host pathogenesis, immune response, and vaccine & treatment updates. J. Gen. Virol. 2021, 102, 001644. [Google Scholar] [CrossRef]
- Corrin, T.; Ackford, R.; Mascarenhas, M.; Greig, J.; Waddell, L.A. Eastern Equine Encephalitis Virus: A Scoping Review of the Global Evidence. Vector Borne Zoonotic Dis. 2021, 21, 305–320. [Google Scholar] [CrossRef]
- Lindsey, N.P.; Staples, J.E.; Fischer, M. Eastern Equine Encephalitis Virus in the United States, 2003–2016. Am. J. Trop. Med. Hyg. 2018, 98, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, P.M.; Andreadis, T.G. Ecology and Epidemiology of Eastern Equine Encephalitis Virus in the Northeastern United States: An Historical Perspective. J. Med. Entomol. 2022, 59, 1–13. [Google Scholar] [CrossRef]
- Schäfer, A.; Brooke, C.B.; Whitmore, A.C.; Johnston, R.E. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J. Virol. 2011, 85, 10682–10690. [Google Scholar] [CrossRef] [Green Version]
- Hollidge, B.S.; Cohen, C.A.; Akuoku Frimpong, J.; Badger, C.V.; Dye, J.M.; Schmaljohn, C.S. Toll-like receptor 4 mediates blood-brain barrier permeability and disease in C3H mice during Venezuelan equine encephalitis virus infection. Virulence 2021, 12, 430–443. [Google Scholar] [CrossRef]
- Bocan, T.M.; Stafford, R.G.; Brown, J.L.; Akuoku Frimpong, J.; Basuli, F.; Hollidge, B.S.; Zhang, X.; Raju, N.; Swenson, R.E.; Smith, D.R. Characterization of Brain Inflammation, Apoptosis, Hypoxia, Blood-Brain Barrier Integrity and Metabolism in Venezuelan Equine Encephalitis Virus (VEEV TC-83) Exposed Mice by In Vivo Positron Emission Tomography Imaging. Viruses 2019, 11, 1052. [Google Scholar] [CrossRef] [Green Version]
- Cain, M.D.; Salimi, H.; Gong, Y.; Yang, L.; Hamilton, S.L.; Heffernan, J.R.; Hou, J.; Miller, M.J.; Klein, R.S. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J. Neuroimmunol. 2017, 308, 118–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, A.T.; Rico, A.B.; Stauft, C.B.; Hammond, S.L.; Aboellail, T.A.; Tjalkens, R.B.; Olson, K.E. Entry Sites of Venezuelan and Western Equine Encephalitis Viruses in the Mouse Central Nervous System following Peripheral Infection. J. Virol. 2016, 90, 5785–5796. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.; Bhattacharya, B.; Puri, R.K.; Maheshwari, R.K. Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genom. 2008, 9, 289. [Google Scholar] [CrossRef] [Green Version]
- Gupta, P.; Sharma, A.; Han, J.; Yang, A.; Bhomia, M.; Knollmann-Ritschel, B.; Puri, R.K.; Maheshwari, R.K. Differential host gene responses from infection with neurovirulent and partially-neurovirulent strains of Venezuelan equine encephalitis virus. BMC Infect. Dis. 2017, 17, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Knollmann-Ritschel, B. Current Understanding of the Molecular Basis of Venezuelan Equine Encephalitis Virus Pathogenesis and Vaccine Development. Viruses 2019, 11, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrera, M.D.; Callahan, V.; Akhrymuk, I.; Bhalla, N.; Zhou, W.; Campbell, C.; Narayanan, A.; Kehn-Hall, K. Proteomic Discovery of VEEV E2-Host Partner Interactions Identifies GRP78 Inhibitor HA15 as a Potential Therapeutic for Alphavirus Infections. Pathogens 2021, 10, 283. [Google Scholar] [CrossRef]
- Carey, B.D.; Bakovic, A.; Callahan, V.; Narayanan, A.; Kehn-Hall, K. New World alphavirus protein interactomes from a therapeutic perspective. Antiviral. Res. 2019, 163, 125–139. [Google Scholar] [CrossRef]
- Yao, Z.; Zanini, F.; Kumar, S.; Karim, M.; Saul, S.; Bhalla, N.; Panpradist, N.; Muniz, A.; Narayanan, A.; Quake, S.R.; et al. The transcriptional landscape of Venezuelan equine encephalitis virus (TC-83) infection. PLoS Negl. Trop. Dis. 2021, 15, e0009306. [Google Scholar] [CrossRef] [PubMed]
- Bakovic, A.; Bhalla, N.; Alem, F.; Campbell, C.; Zhou, W.; Narayanan, A. Inhibitors of Venezuelan Equine Encephalitis Virus Identified Based on Host Interaction Partners of Viral Non-Structural Protein 3. Viruses 2021, 13, 1533. [Google Scholar] [CrossRef] [PubMed]
- Risner, K.; Ahmed, A.; Bakovic, A.; Kortchak, S.; Bhalla, N.; Narayanan, A. Efficacy of FDA-Approved Anti-Inflammatory Drugs Against Venezuelan Equine Encephalitis Virus Infection. Viruses 2019, 11, 1151. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, L.; Brahms, A.; Hooper, I.; Carey, B.; Lin, S.C.; Dahal, B.; Narayanan, A.; Kehn-Hall, K. Repurposed FDA-Approved drug sorafenib reduces replication of Venezuelan equine encephalitis virus and other alphaviruses. Antiviral. Res. 2018, 157, 57–67. [Google Scholar] [CrossRef]
- Amaya, M.; Keck, F.; Lindquist, M.; Voss, K.; Scavone, L.; Kehn-Hall, K.; Roberts, B.; Bailey, C.; Schmaljohn, C.; Narayanan, A. The ubiquitin proteasome system plays a role in venezuelan equine encephalitis virus infection. PLoS ONE 2015, 10, e0124792. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Meertens, L.; Bonazzi, M.; Cossart, P.; Arenzana-Seisdedos, F.; Amara, A. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J. Virol. 2011, 85, 2980–2989. [Google Scholar] [CrossRef] [Green Version]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. mBio 2016, 7, e00804-16. [Google Scholar] [CrossRef] [Green Version]
- Nag, D.K.; Finley, D. A small-molecule inhibitor of deubiquitinating enzyme USP14 inhibits Dengue virus replication. Virus Res. 2012, 165, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Zu, S.; Li, C.; Li, L.; Deng, Y.-Q.; Chen, X.; Luo, D.; Ye, Q.; Huang, Y.-J.; Li, X.-F.; Zhang, R.-R.; et al. TRIM22 suppresses Zika virus replication by targeting NS1 and NS3 for proteasomal degradation. Cell Biosci. 2022, 12, 139. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Pingale, K.D.; Kanade, G.D. Activities of proteasome and m-calpain are essential for Chikungunya virus replication. Virus Genes 2016, 52, 716–721. [Google Scholar] [CrossRef]
- Lee, H.-R.; Lee, M.K.; Kim, C.W.; Kim, M. TRIM Proteins and Their Roles in the Influenza Virus Life Cycle. Microorganisms 2020, 8, 1424. [Google Scholar] [CrossRef]
- Biquand, E.; Poirson, J.; Karim, M.; Declercq, M.; Malausse, N.; Cassonnet, P.; Barbezange, C.; Straub, M.L.; Jones, L.; Munier, S.; et al. Comparative Profiling of Ubiquitin Proteasome System Interplay with Influenza A Virus PB2 Polymerase Protein Recapitulating Virus Evolution in Humans. mSphere 2017, 2, e00330-17. [Google Scholar] [CrossRef] [Green Version]
- Widjaja, I.; de Vries, E.; Tscherne, D.M.; García-Sastre, A.; Rottier, P.J.; de Haan, C.A. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 2010, 84, 9625–9631. [Google Scholar] [CrossRef] [Green Version]
- Park, E.S.; Dezhbord, M.; Lee, A.R.; Kim, K.H. The Roles of Ubiquitination in Pathogenesis of Influenza Virus Infection. Int. J. Mol. Sci. 2022, 23, 4593. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.A.; Berry-Wynne, K.M.; Dos Santos, T.; Addison, W.R.; McFarlane, N.; Hobman, T.; Tyrrell, D.L. HCV and flaviviruses hijack cellular mechanisms for nuclear STAT2 degradation: Up-regulation of PDLIM2 suppresses the innate immune response. PLoS Pathog. 2019, 15, e1007949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; de la Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The Role of IKKβ in Venezuelan Equine Encephalitis Virus Infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef] [PubMed]
- Nenasheva, V.V.; Kovaleva, G.V.; Uryvaev, L.V.; Ionova, K.S.; Dedova, A.V.; Vorkunova, G.K.; Chernyshenko, S.V.; Khaidarova, N.V.; Tarantul, V.Z. Enhanced expression of trim14 gene suppressed Sindbis virus reproduction and modulated the transcription of a large number of genes of innate immunity. Immunol. Res. 2015, 62, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Elbahesh, H. Targeting the proviral host kinase, FAK, limits influenza a virus pathogenesis and NFkB-regulated pro-inflammatory responses. Virology 2019, 534, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Edwards, M.R.; Shabman, R.S.; Leung, D.W.; Endlich-Frazier, A.C.; Borek, D.M.; Otwinowski, Z.; Liu, G.; Huh, J.; Basler, C.F.; et al. Structural basis for Marburg virus VP35-mediated immune evasion mechanisms. Proc. Natl. Acad. Sci. USA 2012, 109, 20661–20666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Basler, C.F. Marburg Virus VP24 Protein Relieves Suppression of the NF-κB Pathway Through Interaction With Kelch-like ECH-Associated Protein 1. J. Infect. Dis. 2015, 212 (Suppl. S2), S154–S159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, M.R.; Johnson, B.; Mire, C.E.; Xu, W.; Shabman, R.S.; Speller, L.N.; Leung, D.W.; Geisbert, T.W.; Amarasinghe, G.K.; Basler, C.F. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep. 2014, 6, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, J.Y.; Chou, Y.Y.; Ciou, J.W.; Liu, F.Y.; Huang, S.P. The Effects of Two Nrf2 Activators, Bardoxolone Methyl and Omaveloxolone, on Retinal Ganglion Cell Survival during Ischemic Optic Neuropathy. Antioxidants 2021, 10, 1466. [Google Scholar] [CrossRef] [PubMed]
- Abed, D.A.; Goldstein, M.; Albanyan, H.; Jin, H.; Hu, L. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein interaction as potential therapeutic and preventive agents. Acta Pharm. Sin. B 2015, 5, 285–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, T.M.; Espina, V.; Lundberg, L.; Pinkham, C.; Brahms, A.; Carey, B.D.; Lin, S.C.; Dahal, B.; Woodson, C.; de la Fuente, C.; et al. Combination Kinase Inhibitor Treatment Suppresses Rift Valley Fever Virus Replication. Viruses 2018, 10, 191. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, L.; Pinkham, C.; de la Fuente, C.; Brahms, A.; Shafagati, N.; Wagstaff, K.M.; Jans, D.A.; Tamir, S.; Kehn-Hall, K. Selective Inhibitor of Nuclear Export (SINE) Compounds Alter New World Alphavirus Capsid Localization and Reduce Viral Replication in Mammalian Cells. PLOS Negl. Trop. Dis. 2016, 10, e0005122. [Google Scholar] [CrossRef] [Green Version]
- Thomas, D.R.; Lundberg, L.; Pinkham, C.; Shechter, S.; DeBono, A.; Baell, J.; Wagstaff, K.M.; Hick, C.A.; Kehn-Hall, K.; Jans, D.A. Identification of novel antivirals inhibiting recognition of Venezuelan equine encephalitis virus capsid protein by the Importin α/β1 heterodimer through high-throughput screening. Antivir. Res. 2018, 151, 8–19. [Google Scholar] [CrossRef]
- Song, M.K.; Lee, J.H.; Ryoo, I.G.; Lee, S.H.; Ku, S.K.; Kwak, M.K. Bardoxolone ameliorates TGF-β1-associated renal fibrosis through Nrf2/Smad7 elevation. Free Radic. Biol. Med. 2019, 138, 33–42. [Google Scholar] [CrossRef]
- Jiang, Z.; Qi, G.; Lu, W.; Wang, H.; Li, D.; Chen, W.; Ding, L.; Yang, X.; Yuan, H.; Zeng, Q. Omaveloxolone inhibits IL-1β-induced chondrocyte apoptosis through the Nrf2/ARE and NF-κB signalling pathways in vitro and attenuates osteoarthritis in vivo. Front. Pharmacol. 2022, 13, 952950. [Google Scholar] [CrossRef]
- Liang, Q.; Dexheimer, T.S.; Zhang, P.; Rosenthal, A.S.; Villamil, M.A.; You, C.; Zhang, Q.; Chen, J.; Ott, C.A.; Sun, H.; et al. A selective USP1-UAF1 inhibitor links deubiquitination to DNA damage responses. Nat. Chem. Biol. 2014, 10, 298–304. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Fan, Y.H.; Xu, X.; Zhang, H.; Dou, J.; Tang, Y.; Zhong, X.; Rojas, Y.; Yu, Y.; Zhao, Y.; et al. A small-molecule inhibitor of UBE2N induces neuroblastoma cell death via activation of p53 and JNK pathways. Cell Death Dis. 2014, 5, e1079. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.Y.; Zhang, Z.H.; Li, R.Z.; Shi, X.K.; Xi, R.Y.; Zhang, G.L.; Li, F.; Wang, F. A small molecule inhibitor of caspase-1 inhibits NLRP3 inflammasome activation and pyroptosis to alleviate gouty inflammation. Immunol. Lett. 2022, 244, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Hodge, C.D.; Edwards, R.A.; Markin, C.J.; McDonald, D.; Pulvino, M.; Huen, M.S.; Zhao, J.; Spyracopoulos, L.; Hendzel, M.J.; Glover, J.N. Covalent Inhibition of Ubc13 Affects Ubiquitin Signaling and Reveals Active Site Elements Important for Targeting. ACS Chem. Biol. 2015, 10, 1718–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Wolf, S.; Beck, B.; Köhler, L.M.; Khoury, K.; Popowicz, G.M.; Goda, S.K.; Subklewe, M.; Twarda, A.; Holak, T.A.; et al. Discovery of highly potent p53-MDM2 antagonists and structural basis for anti-acute myeloid leukemia activities. ACS Chem. Biol. 2014, 9, 802–811. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Suresh Kumar, K.G. The multifaceted roles of USP7: New therapeutic opportunities. Cell Biochem. Biophys. 2011, 60, 61–68. [Google Scholar] [CrossRef]
- Chen, X.; Liu, G.; Yuan, Y.; Wu, G.; Wang, S.; Yuan, L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019, 10, 906. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Bao, C.; Ma, Z.; Xu, B.; Ying, X.; Liu, X.; Zhang, X. Perfluorooctanoic acid stimulates ovarian cancer cell migration, invasion via ERK/NF-κB/MMP-2/-9 pathway. Toxicol. Lett. 2018, 294, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Sha, B.; Huang, W.; Li, M.; Zhao, S.; Zhang, Y.; Yan, J.; Li, Z.; Tang, J.; Duan, P.; et al. ML323, a USP1 inhibitor triggers cell cycle arrest, apoptosis and autophagy in esophageal squamous cell carcinoma cells. Apoptosis 2022, 27, 545–560. [Google Scholar] [CrossRef]
- Park, J.; Cho, J.; Song, E.J. Ubiquitin-proteasome system (UPS) as a target for anticancer treatment. Arch Pharm. Res. 2020, 43, 1144–1161. [Google Scholar] [CrossRef]
- Yuan, T.; Yan, F.; Ying, M.; Cao, J.; He, Q.; Zhu, H.; Yang, B. Inhibition of Ubiquitin-Specific Proteases as a Novel Anticancer Therapeutic Strategy. Front. Pharmacol. 2018, 9, 1080. [Google Scholar] [CrossRef] [Green Version]
- D’Arcy, P.; Wang, X.; Linder, S. Deubiquitinase inhibition as a cancer therapeutic strategy. Pharmacol. Ther. 2015, 147, 32–54. [Google Scholar] [CrossRef] [Green Version]
- Eldridge, A.G.; O’Brien, T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 2010, 17, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, S.; Wu, H. Ubiquitination-Proteasome System (UPS) and Autophagy Two Main Protein Degradation Machineries in Response to Cell Stress. Cells 2022, 11, 851. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, M.I.; Vargas-Cuartas, O.; Gallego-Gomez, J.C.; Shi, P.Y.; Padilla-Sanabria, L.; Castaño-Osorio, J.C.; Rajsbaum, R. K48-linked polyubiquitination of dengue virus NS1 protein inhibits its interaction with the viral partner NS4B. Virus Res. 2018, 246, 1–11. [Google Scholar] [CrossRef]
- Hao, J.; Li, J.; Zhang, Z.; Yang, Y.; Zhou, Q.; Wu, T.; Chen, T.; Wu, Z.; Zhang, P.; Cui, J.; et al. NLRC5 restricts dengue virus infection by promoting the autophagic degradation of viral NS3 through E3 ligase CUL2 (cullin 2). Autophagy 2022. [Google Scholar] [CrossRef]
- van Tol, S.; Kalveram, B.; Ilinykh, P.A.; Ronk, A.; Huang, K.; Aguilera-Aguirre, L.; Bharaj, P.; Hage, A.; Atkins, C.; Giraldo, M.I.; et al. Ubiquitination of Ebola virus VP35 at lysine 309 regulates viral transcription and assembly. PLOS Pathog. 2022, 18, e1010532. [Google Scholar] [CrossRef]
- Shepley-McTaggart, A.; Schwoerer, M.P.; Sagum, C.A.; Bedford, M.T.; Jaladanki, C.K.; Fan, H.; Cassel, J.; Harty, R.N. Ubiquitin Ligase SMURF2 Interacts with Filovirus VP40 and Promotes Egress of VP40 VLPs. Viruses 2021, 13, 288. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Sagum, C.A.; Takizawa, F.; Ruthel, G.; Berry, C.T.; Kong, J.; Sunyer, J.O.; Freedman, B.D.; Bedford, M.T.; Sidhu, S.S.; et al. Ubiquitin Ligase WWP1 Interacts with Ebola Virus VP40 To Regulate Egress. J. Virol. 2017, 91, e00812-17. [Google Scholar] [CrossRef] [Green Version]
- Harty, R.N.; Brown, M.E.; Wang, G.; Huibregtse, J.; Hayes, F.P. A PPxY motif within the VP40 protein of Ebola virus interacts physically and functionally with a ubiquitin ligase: Implications for filovirus budding. Proc. Natl. Acad. Sci. USA 2000, 97, 13871–13876. [Google Scholar] [CrossRef] [Green Version]
- Ramphan, S.; Khongwichit, S.; Saisawang, C.; Kovanich, D.; Ketterman, A.J.; Ubol, S.; Auewarakul, P.; Roytrakul, S.; Smith, D.R.; Kuadkitkan, A. Ubiquitin-Conjugating Enzyme E2 L3 is Downregulated by the Chikungunya Virus nsP2 Protease. Proteomics. Clin. Appl. 2018, 12, e1700020. [Google Scholar] [CrossRef]
- Sun, S.C. Non-canonical NF-κB signaling pathway. Cell Res. 2011, 21, 71–85. [Google Scholar] [CrossRef] [Green Version]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Santoro, M.G.; Rossi, A.; Amici, C. NF-κB and virus infection: Who controls whom. EMBO J. 2003, 22, 2552–2560. [Google Scholar] [CrossRef] [PubMed]
- Yeh, J.X.; Park, E.; Schultz, K.L.W.; Griffin, D.E. NF-κB Activation Promotes Alphavirus Replication in Mature Neurons. J. Virol. 2019, 93, e01071-19. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal. Transduct. Target Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giridharan, S.; Srinivasan, M. Mechanisms of NF-κB p65 and strategies for therapeutic manipulation. J. Inflamm. Res. 2018, 11, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakovic, A.; Bhalla, N.; Kortchak, S.; Sun, C.; Zhou, W.; Ahmed, A.; Risner, K.; Klimstra, W.B.; Narayanan, A. Venezuelan Equine Encephalitis Virus nsP3 Phosphorylation Can Be Mediated by IKKβ Kinase Activity and Abrogation of Phosphorylation Inhibits Negative-Strand Synthesis. Viruses 2020, 12, 1021. [Google Scholar] [CrossRef]
- Keck, F.; Khan, D.; Roberts, B.; Agrawal, N.; Bhalla, N.; Narayanan, A. Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection. Viruses 2018, 10, 606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef] [Green Version]
- Zevini, A.; Ferrari, M.; Olagnier, D.; Hiscott, J. Dengue virus infection and Nrf2 regulation of oxidative stress. Curr. Opin. Virol. 2020, 43, 35–40. [Google Scholar] [CrossRef]
- Ferrari, M.; Zevini, A.; Palermo, E.; Muscolini, M.; Alexandridi, M.; Etna, M.P.; Coccia, E.M.; Fernandez-Sesma, A.; Coyne, C.; Zhang, D.D.; et al. Dengue Virus Targets Nrf2 for NS2B3-Mediated Degradation Leading to Enhanced Oxidative Stress and Viral Replication. J. Virol. 2020, 94, e01551-20. [Google Scholar] [CrossRef]
- Olagnier, D.; Farahani, E.; Thyrsted, J.; Blay-Cadanet, J.; Herengt, A.; Idorn, M.; Hait, A.; Hernaez, B.; Knudsen, A.; Iversen, M.B.; et al. SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun. 2020, 11, 4938. [Google Scholar] [CrossRef]
- Bender, D.; Hildt, E. Effect of Hepatitis Viruses on the Nrf2/Keap1-Signaling Pathway and Its Impact on Viral Replication and Pathogenesis. Int. J. Mol. Sci. 2019, 20, 4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.; Pandey, A.; Manvati, S. Coumarin: An emerging antiviral agent. Heliyon 2020, 6, e03217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sajja, R.K.; Prasad, S.; Tang, S.; Kaisar, M.A.; Cucullo, L. Blood-brain barrier disruption in diabetic mice is linked to Nrf2 signaling deficits: Role of ABCB10? Neurosci. Lett. 2017, 653, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Xi, Z.; Chen, X.; Xu, C.; Wang, B.; Zhong, Z.; Sun, Q.; Sun, Y.; Bian, L. Protocatechuic acid attenuates brain edema and blood-brain barrier disruption after intracerebral hemorrhage in mice by promoting Nrf2/HO-1 pathway. Neuroreport 2020, 31, 1274–1282. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Inhibitor | Abbreviation | Mechanisms and Targets | References |
|---|---|---|---|
| Bardoxolone methyl | BARM | NFkB inhibitor, Nrf2 activator | [41,42,43,47] |
| Bardoxolone | BAR | IKK inhibitor, Nrf2 activator | [41,42,43,47] |
| Omaveloxolone | OMA | NFkB inhibitor, Nrf2 activator | [48,49] |
| NSC697923 | NSC | NFkB inhibitor | [50,51,52] |
| YH239-EE | YH1 | NFkB inhibitor, Nrf2 activator | [53] |
| P005091 | P00 | Ubiquitin-specific protease 7 inhibitor | [54] |
| JSH-23 | JSH | NFkB pathway p65 translocation inhibitor | [55,56] |
| ML-323 | ML | UPS1-UAF1 deubiquitinase complex inhibitor | [49,57] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boghdeh, N.A.; McGraw, B.; Barrera, M.D.; Anderson, C.; Baha, H.; Risner, K.H.; Ogungbe, I.V.; Alem, F.; Narayanan, A. Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses. Viruses 2023, 15, 655. https://doi.org/10.3390/v15030655
Boghdeh NA, McGraw B, Barrera MD, Anderson C, Baha H, Risner KH, Ogungbe IV, Alem F, Narayanan A. Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses. Viruses. 2023; 15(3):655. https://doi.org/10.3390/v15030655
Chicago/Turabian StyleBoghdeh, Niloufar A., Brittany McGraw, Michael D. Barrera, Carol Anderson, Haseebullah Baha, Kenneth H. Risner, Ifedayo V. Ogungbe, Farhang Alem, and Aarthi Narayanan. 2023. "Inhibitors of the Ubiquitin-Mediated Signaling Pathway Exhibit Broad-Spectrum Antiviral Activities against New World Alphaviruses" Viruses 15, no. 3: 655. https://doi.org/10.3390/v15030655