
IL-8 Secreted by Gastric Epithelial Cells Infected with Helicobacter pylori CagA Positive Strains Is a Chemoattractant for Epstein–Barr Virus Infected B Lymphocytes
 , ,
, ,
Abstract
:
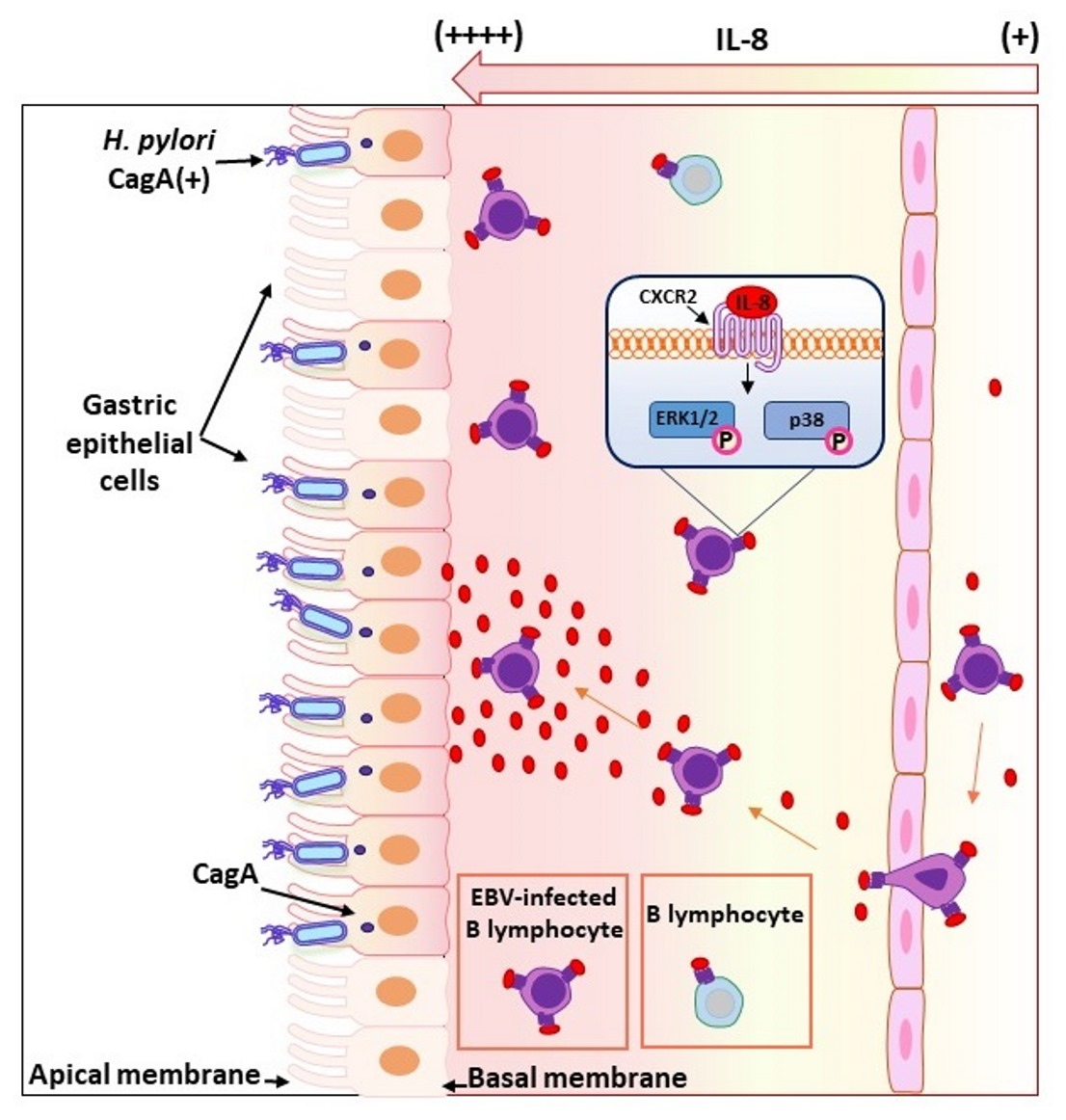{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Helicobacter pylori and Cell Cultures
2.2. Purification of Primary Blood B Lymphocytes (PBLs)
2.3. Infection of Gastric Epithelial Cells and Generation of Conditioned Media
2.3.1. IL-8 Quantification
2.3.2. Chemoattraction Assays: Migration and Invasion
2.4. CagA Overexpression in AGS Cells
Immunofluorescence
2.5. Western Blot Assays
2.6. Quantitative Real-Time PCR Assays
2.7. IL-8 Knockdown
2.8. Bioinformatics Analysis
2.9. Statistical Analysis
3. Results
3.1. EBV-Infected B Lymphocytes Are Chemoattracted by H. pylori CagA(+) Strains
3.2. CagA Overexpression in AGS Cell Promotes the Invasion of EBV-Infected B Lymphocytes
3.3. Active EBV and H. pylori Infection Are Necessary for B Lymphocyte Chemoattraction
3.4. IL-8 Is an Important Chemoattractant for EBV-Infected B Lymphocytes
3.5. CXCR2 Mediates Chemoattraction of EBV-Infected B Lymphocytes in Response to IL-8
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- International Agency for Research on Cancer. Schistosomes, Liver Flukes and Helicobacter Pylori. IARC Monogr. Eval. Carcinog. Risks Hum. 1994, 61, 1–241. [Google Scholar]
- International Agency for Research on Cancer. Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8. IARC Monogr. Eval. Carcinog. Risks Hum. 1997, 70, 1–492. [Google Scholar]
- Marshall, B.J.; Warren, J.R. Unidentified Curved Bacilli in the Stomach of Patients with Gastritis and Peptic Ulceration. Lancet 1984, 1, 1311–1315. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive Molecular Characterization of Gastric Adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitarz, R.; Skierucha, M.; Mielko, J.; Offerhaus, J.; Maciejewski, R.; Polkowski, W. Gastric Cancer: Epidemiology, Prevention, Classification, and Treatment. CMAR 2018, 10, 239–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covacci, A.; Telford, J.L.; Del Giudice, G.; Parsonnet, J.; Rappuoli, R. Helicobacter Pylori Virulence and Genetic Geography. Science 1999, 284, 1328–1333. [Google Scholar] [CrossRef] [Green Version]
- Keates, S.; Keates, A.C.; Warny, M.; Peek, R.M.; Murray, P.G.; Kelly, C.P. Differential Activation of Mitogen-Activated Protein Kinases in AGS Gastric Epithelial Cells by Cag+ and Cag- Helicobacter Pylori. J. Immunol. 1999, 163, 5552–5559. [Google Scholar] [CrossRef]
- Segal, E.D.; Cha, J.; Lo, J.; Falkow, S.; Tompkins, L.S. Altered States: Involvement of Phosphorylated CagA in the Induction of Host Cellular Growth Changes by Helicobacter Pylori. Proc. Natl. Acad. Sci. USA 1999, 96, 14559–14564. [Google Scholar] [CrossRef] [Green Version]
- Higashi, H.; Tsutsumi, R.; Muto, S.; Sugiyama, T.; Azuma, T.; Asaka, M.; Hatakeyama, M. SHP-2 Tyrosine Phosphatase as an Intracellular Target of Helicobacter Pylori CagA Protein. Science 2002, 295, 683–686. [Google Scholar] [CrossRef] [Green Version]
- Segal, E.D.; Falkow, S.; Tompkins, L.S. Helicobacter Pylori Attachment to Gastric Cells Induces Cytoskeletal Rearrangements and Tyrosine Phosphorylation of Host Cell Proteins. Proc. Natl. Acad. Sci. USA 1996, 93, 1259–1264. [Google Scholar] [CrossRef] [Green Version]
- Saadat, I.; Higashi, H.; Obuse, C.; Umeda, M.; Murata-Kamiya, N.; Saito, Y.; Lu, H.; Ohnishi, N.; Azuma, T.; Suzuki, A.; et al. Helicobacter Pylori CagA Targets PAR1/MARK Kinase to Disrupt Epithelial Cell Polarity. Nature 2007, 447, 330–333. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.A.; Tummuru, M.K.; Miller, G.G.; Blaser, M.J. Interleukin-8 Response of Gastric Epithelial Cell Lines to Helicobacter Pylori Stimulation in Vitro. Infect. Immun. 1995, 63, 1681–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, N.; Oomoto, Y.; Mukai, T.; Okamoto, S.; Yamaguchi, S.; Mashiba, H.; Taniyama, K.; Sasaki, N.; Sumii, K.; Haruma, K.; et al. Gastric Corpus IL-8 Concentration and Neutrophil Infiltration in Duodenal Ulcer Patients. Aliment. Pharmacol. Ther. 1997, 11, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Kashima, K.; Imanishi, J. Induction of Various Cytokines and Development of Severe Mucosal Inflammation by CagA Gene Positive Helicobacter Pylori Strains. Gut 1997, 41, 442–451. [Google Scholar] [CrossRef] [Green Version]
- Camorlinga-Ponce, M.; Aviles-Jimenez, F.; Cabrera, L.; Hernández-Pando, R.; Muñoz, O.; Soza, J.; Torres, J. Intensity of Inflammation, Density of Colonization and Interleukin-8 Response in the Gastric Mucosa of Children Infected with Helicobacter Pylori. Helicobacter 2003, 8, 554–560. [Google Scholar] [CrossRef]
- Knall, C.; Young, S.; Nick, J.A.; Buhl, A.M.; Worthen, G.S.; Johnson, G.L. Interleukin-8 Regulation of the Ras/Raf/Mitogen-Activated Protein Kinase Pathway in Human Neutrophils. J. Biol. Chem. 1996, 271, 2832–2838. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wang, Q.; Chen, B.; Zhao, Y.; Shen, B.; Wang, H.; Xu, J.; Zhu, M.; Zhao, X.; Xu, C.; et al. Gastric Cancer Mesenchymal Stem Cells Derived IL-8 Induces PD-L1 Expression in Gastric Cancer Cells via STAT3/MTOR-c-Myc Signal Axis. Cell Death Dis. 2018, 9, 928. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence–Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [CrossRef] [Green Version]
- Vincent-Bugnas, S.; Vitale, S.; Mouline, C.C.; Khaali, W.; Charbit, Y.; Mahler, P.; Prêcheur, I.; Hofman, P.; Maryanski, J.L.; Doglio, A. EBV Infection Is Common in Gingival Epithelial Cells of the Periodontium and Worsens during Chronic Periodontitis. PLoS ONE 2013, 8, e80336. [Google Scholar] [CrossRef] [Green Version]
- Hudnall, S.D.; Ge, Y.; Wei, L.; Yang, N.-P.; Wang, H.-Q.; Chen, T. Distribution and Phenotype of Epstein-Barr Virus-Infected Cells in Human Pharyngeal Tonsils. Mod. Pathol. 2005, 18, 519–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tugizov, S.M.; Berline, J.W.; Palefsky, J.M. Epstein-Barr Virus Infection of Polarized Tongue and Nasopharyngeal Epithelial Cells. Nat. Med. 2003, 9, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Walling, D.M.; Flaitz, C.M.; Nichols, C.M.; Hudnall, S.D.; Adler-Storthz, K. Persistent Productive Epstein-Barr Virus Replication in Normal Epithelial Cells in Vivo. J. Infect. Dis. 2001, 184, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The Dynamics of EBV Shedding Implicate a Central Role for Epithelial Cells in Amplifying Viral Output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [Green Version]
- Joseph, A.M.; Babcock, G.J.; Thorley-Lawson, D.A. Cells Expressing the Epstein-Barr Virus Growth Program Are Present in and Restricted to the Naive B-Cell Subset of Healthy Tonsils. J. Virol. 2000, 74, 9964–9971. [Google Scholar] [CrossRef] [Green Version]
- Chaganti, S.; Heath, E.M.; Bergler, W.; Kuo, M.; Buettner, M.; Niedobitek, G.; Rickinson, A.B.; Bell, A.I. Epstein-Barr Virus Colonization of Tonsillar and Peripheral Blood B-Cell Subsets in Primary Infection and Persistence. Blood 2009, 113, 6372–6381. [Google Scholar] [CrossRef]
- Cárdenas-Mondragón, M.G.; Torres, J.; Sánchez-Zauco, N.; Gómez-Delgado, A.; Camorlinga-Ponce, M.; Maldonado-Bernal, C.; Fuentes-Pananá, E.M. Elevated Levels of Interferon-γ Are Associated with High Levels of Epstein-Barr Virus Reactivation in Patients with the Intestinal Type of Gastric Cancer. J. Immunol. Res. 2017, 2017, 7069242. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas-Mondragón, M.G.; Torres, J.; Flores-Luna, L.; Carreón-Talavera, R.; Camorlinga-Ponce, M.; Fuentes-Pananá, E.M. Epstein-Barr Virus Association with Peptic Ulcer Disease. Anal. Cell. Pathol. 2015, 2015, 164840. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas-Mondragón, M.G.; Torres, J.; Flores-Luna, L.; Camorlinga-Ponce, M.; Carreón-Talavera, R.; Gomez-Delgado, A.; Kasamatsu, E.; Fuentes-Pananá, E.M. Case–Control Study of Epstein–Barr Virus and Helicobacter Pylori Serology in Latin American Patients with Gastric Disease. Br. J. Cancer 2015, 112, 1866–1873. [Google Scholar] [CrossRef] [Green Version]
- Cárdenas-Mondragón, M.G.; Carreón-Talavera, R.; Camorlinga-Ponce, M.; Gomez-Delgado, A.; Torres, J.; Fuentes-Pananá, E.M. Epstein Barr Virus and Helicobacter Pylori Co-Infection Are Positively Associated with Severe Gastritis in Pediatric Patients. PLoS ONE 2013, 8, e62850. [Google Scholar] [CrossRef] [Green Version]
- Morales-Sánchez, A.; Torres, J.; Cardenas-Mondragón, M.G.; Romo-González, C.; Camorlinga-Ponce, M.; Flores-Luna, L.; Fuentes-Pananá, E.M. Detection of Epstein-Barr Virus DNA in Gastric Biopsies of Pediatric Patients with Dyspepsia. Pathogens 2020, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Leon, A.; Atherton, J.C.; Argent, R.H.; Puente, J.L.; Torres, J. Heterogeneity in the Activity of Mexican Helicobacter Pylori Strains in Gastric Epithelial Cells and Its Association with Diversity in the CagA Gene. Infect. Immun. 2007, 75, 3445–3454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maruo, S.; Yang, L.; Takada, K. Roles of Epstein-Barr Virus Glycoproteins Gp350 and Gp25 in the Infection of Human Epithelial Cells. J. Gen. Virol. 2001, 82, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- Bagnoli, F.; Buti, L.; Tompkins, L.; Covacci, A.; Amieva, M.R. Helicobacter Pylori CagA Induces a Transition from Polarized to Invasive Phenotypes in MDCK Cells. Proc. Natl. Acad. Sci. USA 2005, 102, 16339–16344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From Reads to Genes to Pathways: Differential Expression Analysis of RNA-Seq Experiments Using Rsubread and the EdgeR Quasi-Likelihood Pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Duncan, D.; Shi, Z.; Zhang, B. WEB-Based GEne SeT AnaLysis Toolkit (WebGestalt): Update 2013. Nucleic Acids Res. 2013, 41, W77–W83. [Google Scholar] [CrossRef] [Green Version]
- Cheng, K.-S.; Tang, H.-L.; Chou, F.-T. Serum IL-8 as a Possible Marker for Determining the Status of Helicobacter Pylori Infection in Patients with Untreated and Treated Peptic Ulcer. Adv. Ther. 2004, 21, 39–46. [Google Scholar] [CrossRef]
- Li, X.; Zhai, J.; Shen, Y.; Zhang, T.; Wang, Y.; He, Y.; You, Q.; Shen, L. Tumor-Derived IL-8 Facilitates Lymph Node Metastasis of Gastric Cancer via PD-1 up-Regulation in CD8+ T Cells. Cancer Immunol. Immunother. 2022, 71, 3057–3070. [Google Scholar] [CrossRef]
- Crabtree, J.E.; Covacci, A.; Farmery, S.M.; Xiang, Z.; Tompkins, D.S.; Perry, S.; Lindley, I.J.; Rappuoli, R. Helicobacter Pylori Induced Interleukin-8 Expression in Gastric Epithelial Cells Is Associated with CagA Positive Phenotype. J. Clin. Pathol. 1995, 48, 41–45. [Google Scholar] [CrossRef]
- Allison, C.C.; Ferrand, J.; McLeod, L.; Hassan, M.; Kaparakis-Liaskos, M.; Grubman, A.; Bhathal, P.S.; Dev, A.; Sievert, W.; Jenkins, B.J.; et al. Nucleotide Oligomerization Domain 1 Enhances IFN-γ Signaling in Gastric Epithelial Cells during Helicobacter Pylori Infection and Exacerbates Disease Severity. J. Immunol. 2013, 190, 3706–3715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cook, K.W.; Letley, D.P.; Ingram, R.J.M.; Staples, E.; Skjoldmose, H.; Atherton, J.C.; Robinson, K. CCL20/CCR6-Mediated Migration of Regulatory T Cells to the Helicobacter Pylori-Infected Human Gastric Mucosa. Gut 2014, 63, 1550–1559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebrell, T.A.; Hashimi, M.; Sidar, B.; Wilkinson, R.A.; Kirpotina, L.; Quinn, M.T.; Malkoç, Z.; Taylor, P.J.; Wilking, J.N.; Bimczok, D. A Novel Gastric Spheroid Co-Culture Model Reveals Chemokine-Dependent Recruitment of Human Dendritic Cells to the Gastric Epithelium. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 157–171.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikuse, T.; Ohtsuka, Y.; Kudo, T.; Hosoi, K.; Obayashi, N.; Jimbo, K.; Aoyagi, Y.; Fujii, T.; Nagata, S.; Shimizu, T. Microarray Analysis of Gastric Mucosa among Children with Helicobacter Pylori Infection. Pediatr. Int. 2012, 54, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Mejías-Luque, R.; Zöller, J.; Anderl, F.; Loew-Gil, E.; Vieth, M.; Adler, T.; Engler, D.B.; Urban, S.; Browning, J.L.; Müller, A.; et al. Lymphotoxin β Receptor Signalling Executes Helicobacter Pylori-Driven Gastric Inflammation in a T4SS-Dependent Manner. Gut 2017, 66, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.-S.; Zhao, Y.-L.; Li, M.-S.; Liu, Y.-G.; Cheng, P.; Lv, Y.-P.; Mao, F.-Y.; Chen, W.; Yang, S.-M.; Hao, C.-J.; et al. Upexpression of BHLHE40 in Gastric Epithelial Cells Increases CXCL12 Production through Interaction with P-STAT3 in Helicobacter Pylori-Associated Gastritis. FASEB J. 2020, 34, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaiboullina, S.F.; Abdulkhakov, S.; Khalikova, A.; Safina, D.; Martynova, E.V.; Davidyuk, Y.; Khuzin, F.; Faizullina, R.; Lombardi, V.C.; Cherepnev, G.V.; et al. Serum Cytokine Signature That Discriminates Helicobacter Pylori Positive and Negative Juvenile Gastroduodenitis. Front. Microbiol. 2016, 7, 1916. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, A.; Isomoto, H.; Hisatsune, J.; Nakayama, M.; Nakashima, Y.; Matsushima, K.; Mizuta, Y.; Hayashi, T.; Yamaoka, Y.; Azuma, T.; et al. Enhanced Expression of CCL20 in Human Helicobacter Pylori-Associated Gastritis. Clin. Immunol. 2009, 3, 290–297. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, K.; Nishiumi, S.; Yang, L.; Klimatcheva, E.; Pandina, T.; Takahashi, S.; Matsui, H.; Nakamura, M.; Zauderer, M.; Yoshida, M.; et al. Anti-CXCL13 Antibody Can Inhibit the Formation of Gastric Lymphoid Follicles Induced by Helicobacter Infection. Mucosal Immunol. 2014, 7, 1244–1254. [Google Scholar] [CrossRef] [Green Version]
- Obayashi, N.; Ohtsuka, Y.; Hosoi, K.; Ikuse, T.; Jimbo, K.; Aoyagi, Y.; Fujii, T.; Kudo, T.; Asaoka, D.; Hojo, M.; et al. Comparison of Gene Expression Between Pediatric and Adult Gastric Mucosa with Helicobacter Pylori Infection. Helicobacter 2016, 21, 114–123. [Google Scholar] [CrossRef]
- Nakashima, Y.; Isomoto, H.; Matsushima, K.; Yoshida, A.; Nakayama, T.; Nakayama, M.; Hisatsune, J.; Ichikawa, T.; Takeshima, F.; Hayashi, T.; et al. Enhanced Expression of CXCL13 in Human Helicobacter Pylori-Associated Gastritis. Dig. Dis. Sci. 2011, 56, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-Y.; Tsai, H.-F.; Lin, W.-C.; Hsu, P.-I.; Shun, C.-T.; Wu, M.-S.; Hsu, P.-N. Upregulation of CCL20 and Recruitment of CCR6+ Gastric Infiltrating Lymphocytes in Helicobacter Pylori Gastritis. Infect. Immun. 2007, 75, 4357–4363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, F.-Y.; Lv, Y.-P.; Hao, C.-J.; Teng, Y.-S.; Liu, Y.-G.; Cheng, P.; Yang, S.-M.; Chen, W.; Liu, T.; Zou, Q.-M.; et al. Helicobacter Pylori-Induced Rev-Erbα Fosters Gastric Bacteria Colonization by Impairing Host Innate and Adaptive Defense. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 395–425. [Google Scholar] [CrossRef] [PubMed]
- Ming, S.; Yin, H.; Li, X.; Gong, S.; Zhang, G.; Wu, Y. GITR Promotes the Polarization of TFH-Like Cells in Helicobacter Pylori-Positive Gastritis. Front. Immunol. 2021, 12, 736269. [Google Scholar] [CrossRef] [PubMed]
- Planet, E. PhenoTest: Tools to Test Association between Gene Expression and Phenotype in a Way That Is Efficient, Structured, Fast and Scalable. We Also Provide Tools to Do GSEA (Gene Set Enrichment Analysis) and Copy Number Variation; R Package Version 1.46.0; CRAN: Vienna, Austria, 2022. [Google Scholar]
- Therneau, T.M.; Grambsch, P.M. Modeling Survival Data: Extending the Cox Model; Dietz, K., Gail, M., Krickeberg, K., Samet, J., Tsiatis, A., Eds.; Statistics for Biology and Health; Springer: New York, NY, USA, 2000; ISBN 978-1-4419-3161-0. [Google Scholar]
- Grolemund, G.; Wickham, H. Dates and Times Made Easy with Lubridate. J. Stat. Soft. 2011, 40, 1–25. [Google Scholar] [CrossRef]
- Yamaoka, Y.; Kita, M.; Kodama, T.; Sawai, N.; Tanahashi, T.; Kashima, K.; Imanishi, J. Chemokines in the Gastric Mucosa in Helicobacter Pylori Infection. Gut 1998, 42, 609–617. [Google Scholar] [CrossRef] [Green Version]
- Rieder, G.; Einsiedl, W.; Hatz, R.A.; Stolte, M.; Enders, G.A.; Walz, A. Comparison of CXC Chemokines ENA-78 and Interleukin-8 Expression in Helicobacter Pylori-Associated Gastritis. Infect. Immun. 2001, 69, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Qu, N.; Peng, J.; Yue, C.; Yuan, L.; Yuan, Y. CagA Promotes Proliferation and Inhibits Apoptosis of GES-1 Cells by Upregulating TRAF1/4-1BB. Mol. Med. Rep. 2017, 16, 1262–1268. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.-Y.; Lee, Y.-C.; Kim, H.K.; Blaser, M.J. Helicobacter Pylori CagA Transfection of Gastric Epithelial Cells Induces Interleukin-8. Cell. Microbiol. 2006, 8, 97–106. [Google Scholar] [CrossRef]
- Holmes, W.E.; Lee, J.; Kuang, W.J.; Rice, G.C.; Wood, W.I. Structure and Functional Expression of a Human Interleukin-8 Receptor. Science 1991, 253, 1278–1280. [Google Scholar] [CrossRef]
- Murphy, P.M.; Tiffany, H.L. Cloning of Complementary DNA Encoding a Functional Human Interleukin-8 Receptor. Science 1991, 253, 1280–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein-Barr Virus Reprograms Human B Lymphocytes Immediately in the Prelatent Phase of Infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Z.; Xia, G.; Xiang, Z.; Liu, M.; Wei, Z.; Yan, J.; Chen, W.; Zhu, J.; Awasthi, N.; Sun, X.; et al. A C-X-C Chemokine Receptor Type 2–Dominated Cross-Talk between Tumor Cells and Macrophages Drives Gastric Cancer Metastasis. Clin. Cancer Res. 2019, 25, 3317–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zarbock, A.; Allegretti, M.; Ley, K. Therapeutic Inhibition of CXCR2 by Reparixin Attenuates Acute Lung Injury in Mice. Br. J. Pharmacol. 2008, 155, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Bertini, R.; Allegretti, M.; Bizzarri, C.; Moriconi, A.; Locati, M.; Zampella, G.; Cervellera, M.N.; Di Cioccio, V.; Cesta, M.C.; Galliera, E.; et al. Noncompetitive Allosteric Inhibitors of the Inflammatory Chemokine Receptors CXCR1 and CXCR2: Prevention of Reperfusion Injury. Proc. Natl. Acad. Sci. USA 2004, 101, 11791–11796. [Google Scholar] [CrossRef] [Green Version]
- Martín Guerrero, J.M.; Hergueta Delgado, P.; Esteban Carretero, J.J.; Rivera Hueto, F.F.; Pellicer Bautista, F.J.; Herrerías Gutiérrez, J.M. Pathogenic Implications of Interleukin-8 Activity and Bacterial Phenotype in Antral Gastritis Associated with Helicobacter Pylori. Rev. Esp. Enferm. Dig. 2000, 92, 301–315. [Google Scholar]
- Thorley-Lawson, D.A.; Hawkins, J.B.; Tracy, S.I.; Shapiro, M. The Pathogenesis of Epstein–Barr Virus Persistent Infection. Curr. Opin. Virol. 2013, 3, 227–232. [Google Scholar] [CrossRef] [Green Version]
- Louissaint, A.; Ferry, J.A.; Soupir, C.P.; Hasserjian, R.P.; Harris, N.L.; Zukerberg, L.R. Infectious Mononucleosis Mimicking Lymphoma: Distinguishing Morphological and Immunophenotypic Features. Mod. Pathol. 2012, 25, 1149–1159. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.-W.; Chang, S.-T.; Lu, C.-L.; Hwang, W.-S.; Tsao, C.-J.; Huang, W.-T.; Chang, K.-Y.; Chuang, S.-S. Upper Aerodigestive Tract Lymphoma in Taiwan. J. Clin. Pathol. 2010, 63, 888–893. [Google Scholar] [CrossRef]
- Kojima, M.; Nakamura, N.; Itoh, H.; Shimizu, K.; Shimizu, K.; Matsuda, H.; Tamaki, Y.; Masawa, N.; Nakamura, S. Epstein-Barr Virus-Related Atypical Lymphoproliferative Disorders in Waldeyer’s Ring: A Clinicopathological Study of 9 Cases. Pathobiology 2010, 77, 218–224. [Google Scholar] [CrossRef]
- Wada, T.; Muraoka, M.; Yokoyama, T.; Toma, T.; Kanegane, H.; Yachie, A. Cytokine Profiles in Children with Primary Epstein-Barr Virus Infection. Pediatr. Blood Cancer 2013, 60, E46–E48. [Google Scholar] [CrossRef] [Green Version]
- Martínez-López, J.; Torres, J.; Camorlinga-Ponce, M.; Mantilla, A.; Leal, Y.; Fuentes-Pananá, E. Evidence of Epstein-Barr Virus Association with Gastric Cancer and Non-Atrophic Gastritis. Viruses 2014, 6, 301–318. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Bae, B.-N.; Kang, G.; Kim, H.-J.; Park, K. Cytokine Expression Associated with Helicobacter Pylori and Epstein-Barr Virus Infection in Gastric Carcinogenesis. APMIS 2017, 125, 808–815. [Google Scholar] [CrossRef]
- Saju, P.; Murata-Kamiya, N.; Hayashi, T.; Senda, Y.; Nagase, L.; Noda, S.; Matsusaka, K.; Funata, S.; Kunita, A.; Urabe, M.; et al. Host SHP1 Phosphatase Antagonizes Helicobacter Pylori CagA and Can Be Downregulated by Epstein–Barr Virus. Nat. Microbiol. 2016, 1, 16026. [Google Scholar] [CrossRef]
- Wu, W.K.; Yu, J.; Chan, M.T.; To, K.F.; Cheng, A.S. Combinatorial Epigenetic Deregulation by Helicobacter Pylori and Epstein-Barr Virus Infections in Gastric Tumourigenesis. J. Pathol. 2016, 239, 245–249. [Google Scholar] [CrossRef] [Green Version]
- Zong, L.; Seto, Y. CpG Island Methylator Phenotype, Helicobacter Pylori, Epstein-Barr Virus, and Microsatellite Instability and Prognosis in Gastric Cancer: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 9, e86097. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Jha, H.C.; Shukla, S.K.; Shirley, M.K.; Robertson, E.S. Epigenetic Regulation of Tumor Suppressors by Helicobacter pylori Enhances EBV-Induced Proliferation of Gastric Epithelial Cells. mBio 2018, 9, e00649-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, A.; Nath Prasad, K.; Chand Ghoshal, U.; Krishnani, N.; Roshan Bhagat, M.; Husain, N. Association of Helicobacter Pylori and Epstein-Barr Virus with Gastric Cancer and Peptic Ulcer Disease. Scand. J. Gastroenterol. 2008, 43, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Minoura-Etoh, J.; Gotoh, K.; Sato, R.; Ogata, M.; Kaku, N.; Fujioka, T.; Nishizono, A. Helicobacter Pylori-Associated Oxidant Monochloramine Induces Reactivation of Epstein-Barr Virus (EBV) in Gastric Epithelial Cells Latently Infected with EBV. J. Med. Microbiol. 2006, 55, 905–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Végran, F.; Boidot, R.; Michiels, C.; Sonveaux, P.; Feron, O. Lactate Influx through the Endothelial Cell Monocarboxylate Transporter MCT1 Supports an NF-ΚB/IL-8 Pathway That Drives Tumor Angiogenesis. Cancer Res. 2011, 71, 2550–2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Wei, P.-K. Interleukin-8: A Potent Promoter of Angiogenesis in Gastric Cancer. Oncol. Lett. 2016, 11, 1043–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.; He, H.; Liu, H.; Li, R.; Chen, Y.; Qi, Y.; Jiang, Q.; Chen, L.; Zhang, P.; Zhang, H.; et al. Tumour-Associated Macrophages-Derived CXCL8 Determines Immune Evasion through Autonomous PD-L1 Expression in Gastric Cancer. Gut 2019, 68, 1764–1773. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, S.; Lina, T.T.; Gonzalez, J.; Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Effect of Helicobacter Pylori on Gastric Epithelial Cells. World J. Gastroenterol. 2014, 20, 12767–12780. [Google Scholar] [CrossRef] [PubMed]
- Augusto, A.C.; Miguel, F.; Mendonça, S.; Pedrazzoli, J.; Gurgueira, S.A. Oxidative Stress Expression Status Associated to Helicobacter Pylori Virulence in Gastric Diseases. Clin. Biochem. 2007, 40, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Davies, G.R.; Simmonds, N.J.; Stevens, T.R.; Sheaff, M.T.; Banatvala, N.; Laurenson, I.F.; Blake, D.R.; Rampton, D.S. Helicobacter Pylori Stimulates Antral Mucosal Reactive Oxygen Metabolite Production in Vivo. Gut 1994, 35, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.F.; Genta, R.M.; Yardley, J.H.; Correa, P. Classification and Grading of Gastritis. The Updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am. J. Surg. Pathol. 1996, 20, 1161–1181. [Google Scholar] [CrossRef]
- Slots, J. Periodontal Herpesviruses: Prevalence, Pathogenicity, Systemic Risk. Periodontol. 2000 2015, 69, 28–45. [Google Scholar] [CrossRef]
- Slots, J.; Slots, H. Periodontal Herpesvirus Morbidity and Treatment. Periodontol. 2000 2019, 79, 210–220. [Google Scholar] [CrossRef]
- Yee, M.; Kim, S.; Sethi, P.; Düzgüneş, N.; Konopka, K. Porphyromonas Gingivalis Stimulates IL-6 and IL-8 Secretion in GMSMK, HSC-3 and H413 Oral Epithelial Cells. Anaerobe 2014, 28, 62–67. [Google Scholar] [CrossRef]
- Sandros, J.; Karlsson, C.; Lappin, D.F.; Madianos, P.N.; Kinane, D.F.; Papapanou, P.N. Cytokine Responses of Oral Epithelial Cells to Porphyromonas Gingivalis Infection. J. Dent. Res. 2000, 79, 1808–1814. [Google Scholar] [CrossRef]
- Guentsch, A.; Rönnebeck, M.; Puklo, M.; Preshaw, P.M.; Pfister, W.; Eick, S. Influence of Serum on Interaction of Porphyromonas Gingivalis ATCC 33277 and Aggregatibacter Actinomycetemcomitans Y4 with an Epithelial Cell Line. J. Periodontal Res. 2010, 45, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.T.-J.; Haake, S.K.; Park, N.-H. Gingival Epithelial Cells Increase Interleukin-8 Secretion in Response to Actinobacillus Actinomycetemcomitans Challenge. J. Periodontol. 1998, 69, 1105–1110. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Moffatt, C.E.; Hagerty, D.; Whitmore, S.E.; Brown, T.A.; Graves, D.T.; Lamont, R.J. Interaction of Oral Bacteria with Gingival Epithelial Cell Multilayers: Gingival Epithelial Multilayer Responses to Bacteria. Mol. Oral Microbiol. 2011, 26, 210–220. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-P.; Chan, A.T.C.; Le, Q.-T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal Carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Nasr, H.B.; Chahed, K.; Mestiri, S.; Bouaouina, N.; Snoussi, K.; Chouchane, L. Association of IL-8 (−251)T/A Polymorphism with Susceptibility to and Aggressiveness of Nasopharyngeal Carcinoma. Hum. Immunol. 2007, 68, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.-S.; Lan, Y.; Tang, R.-G.; Xu, Q.-Q.; Huang, Y.; Nong, H.-B.; Huang, W.-T. Single Nucleotide Polymorphism and Haplotype Association of the Interleukin-8 Gene with Nasopharyngeal Carcinoma. Clin. Immunol. 2007, 125, 309–317. [Google Scholar] [CrossRef]
- Jones, S.A.; Wolf, M.; Qin, S.; Mackay, C.R.; Baggiolini, M. Different Functions for the Interleukin 8 Receptors (IL-8R) of Human Neutrophil Leukocytes: NADPH Oxidase and Phospholipase D Are Activated through IL-8R1 but Not IL-8R2. Proc. Natl. Acad. Sci. USA 1996, 93, 6682–6686. [Google Scholar] [CrossRef] [Green Version]
- Martínez Muñoz, L.; Lucas, P.; Navarro, G.; Checa, A.I.; Franco, R.; Martínez-A, C.; Rodríguez-Frade, J.M.; Mellado, M. Dynamic Regulation of CXCR1 and CXCR2 Homo- and Heterodimers. J. Immunol. 2009, 183, 7337–7346. [Google Scholar] [CrossRef] [Green Version]
- Korbecki, J.; Kupnicka, P.; Chlubek, M.; Gorący, J.; Gutowska, I.; Baranowska-Bosiacka, I. CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer. Int. J. Mol. Sci. 2022, 23, 2168. [Google Scholar] [CrossRef]
- Li, Z.; Wang, Y.; Dong, S.; Ge, C.; Xiao, Y.; Li, R.; Ma, X.; Xue, Y.; Zhang, Q.; Lv, J.; et al. Association of CXCR1 and 2 Expressions with Gastric Cancer Metastasis in Ex Vivo and Tumor Cell Invasion in Vitro. Cytokine 2014, 69, 6–13. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, H.; Shen, Z.; Wang, X.; Zhang, H.; Qin, J.; Xu, J.; Sun, Y.; Qin, X. The Prognostic Value of CXC-Chemokine Receptor 2 (CXCR2) in Gastric Cancer Patients. BMC Cancer 2015, 15, 766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waugh, D.J.J.; Wilson, C. The Interleukin-8 Pathway in Cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Domínguez-Martínez, D.A.; Fontes-Lemus, J.I.; García-Regalado, A.; Juárez-Flores, Á.; Fuentes-Pananá, E.M. IL-8 Secreted by Gastric Epithelial Cells Infected with Helicobacter pylori CagA Positive Strains Is a Chemoattractant for Epstein–Barr Virus Infected B Lymphocytes. Viruses 2023, 15, 651. https://doi.org/10.3390/v15030651
Domínguez-Martínez DA, Fontes-Lemus JI, García-Regalado A, Juárez-Flores Á, Fuentes-Pananá EM. IL-8 Secreted by Gastric Epithelial Cells Infected with Helicobacter pylori CagA Positive Strains Is a Chemoattractant for Epstein–Barr Virus Infected B Lymphocytes. Viruses. 2023; 15(3):651. https://doi.org/10.3390/v15030651
Chicago/Turabian StyleDomínguez-Martínez, Diana A., José I. Fontes-Lemus, Alejandro García-Regalado, Ángel Juárez-Flores, and Ezequiel M. Fuentes-Pananá. 2023. "IL-8 Secreted by Gastric Epithelial Cells Infected with Helicobacter pylori CagA Positive Strains Is a Chemoattractant for Epstein–Barr Virus Infected B Lymphocytes" Viruses 15, no. 3: 651. https://doi.org/10.3390/v15030651