
Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution
 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. JEV Analysis Pipeline Design
2.2. Dataset Construction in the Analyses
2.3. Time Scaled Phylogenetic Analysis of the JEV
2.4. Analysis of the Geographic Migration of JEV GIV
2.5. Phylogenetic Analysis Based on the Variable Region within 3’UTRs of JEVs
2.6. Sequence Analysis of GIV JEVs
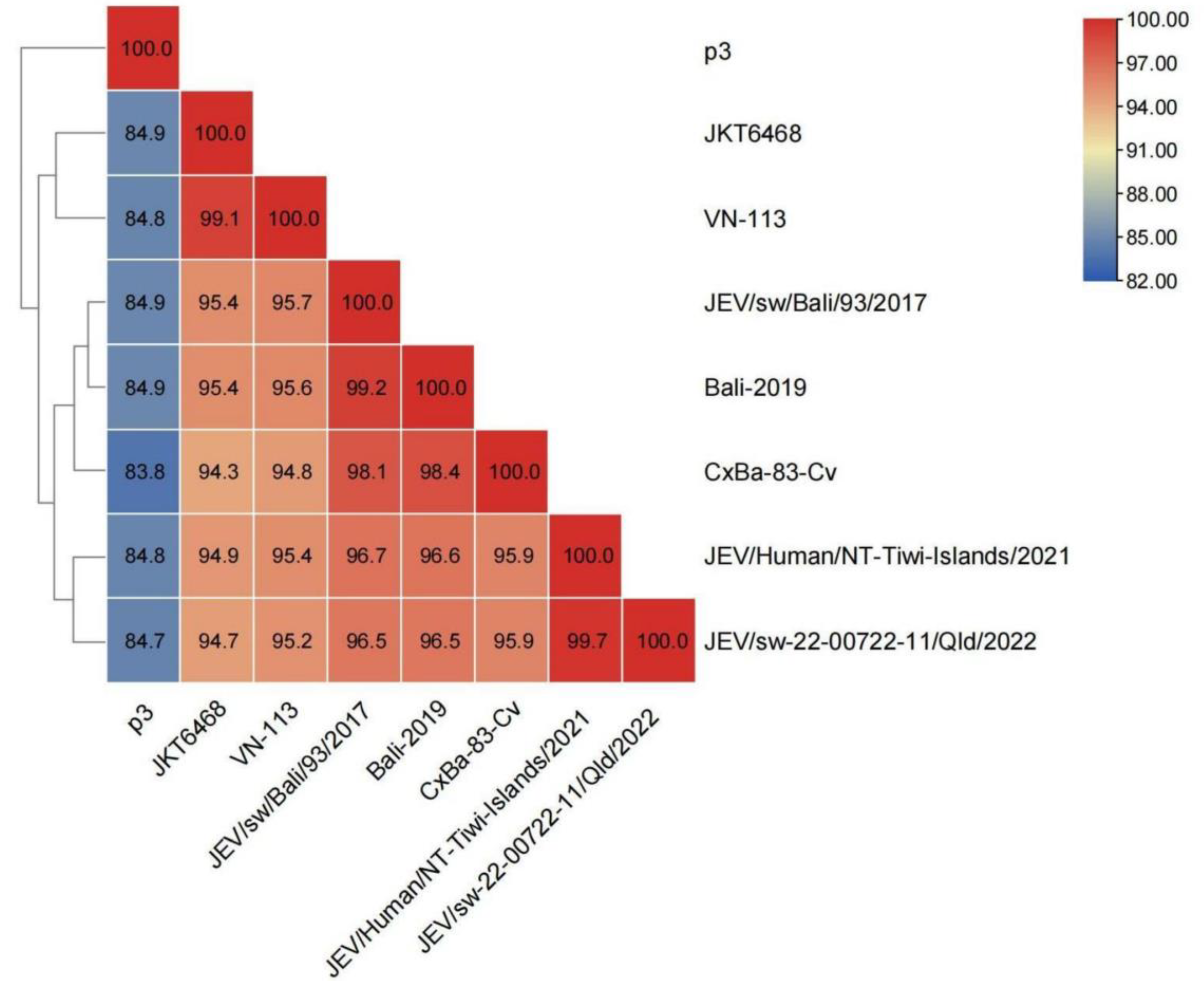
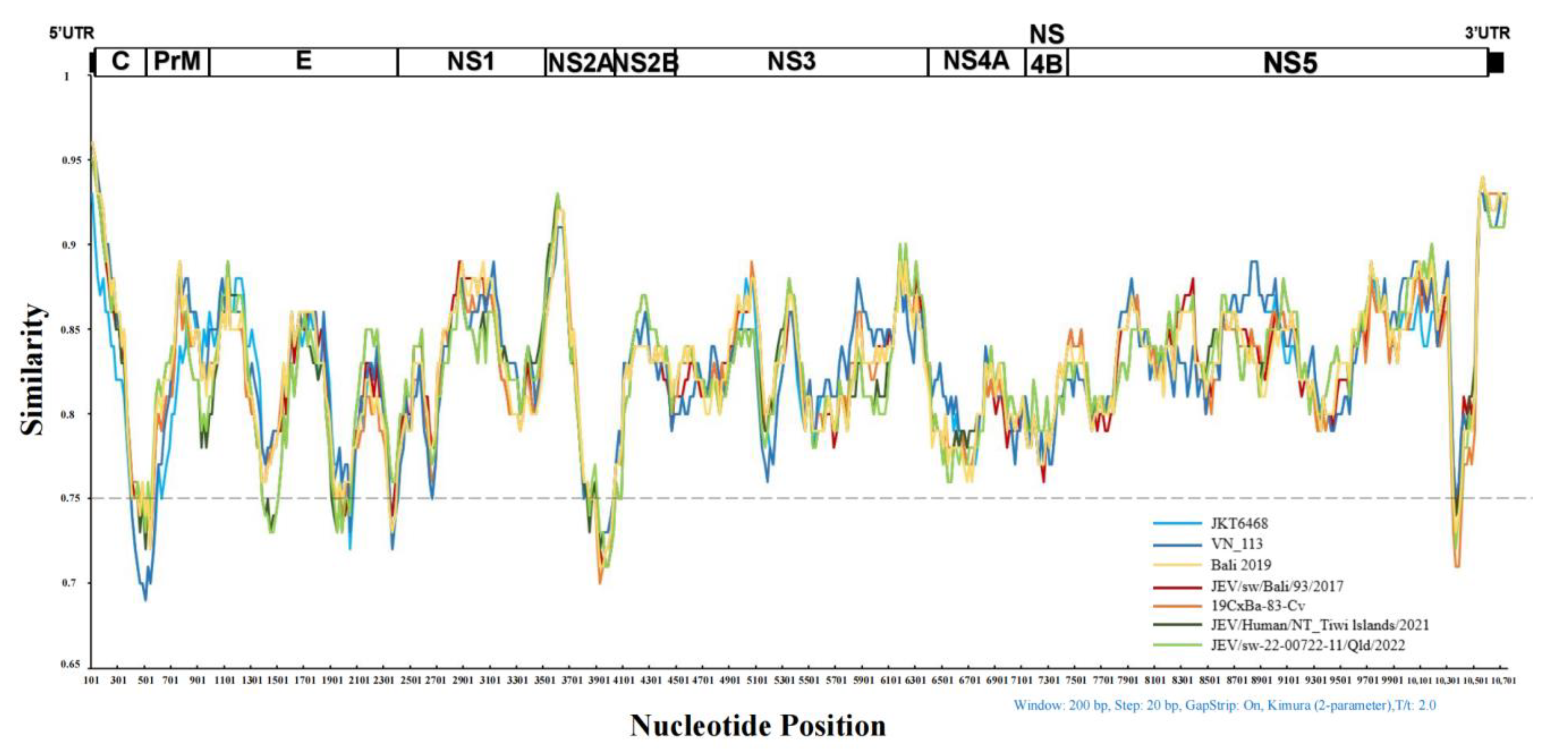
2.6.1. Analysis of the Whole Genome Sequence Similarity of GIV JEVs
2.6.2. Analysis of the Nucleotide and Amino Acid Similarity in Different JEV Genotypes
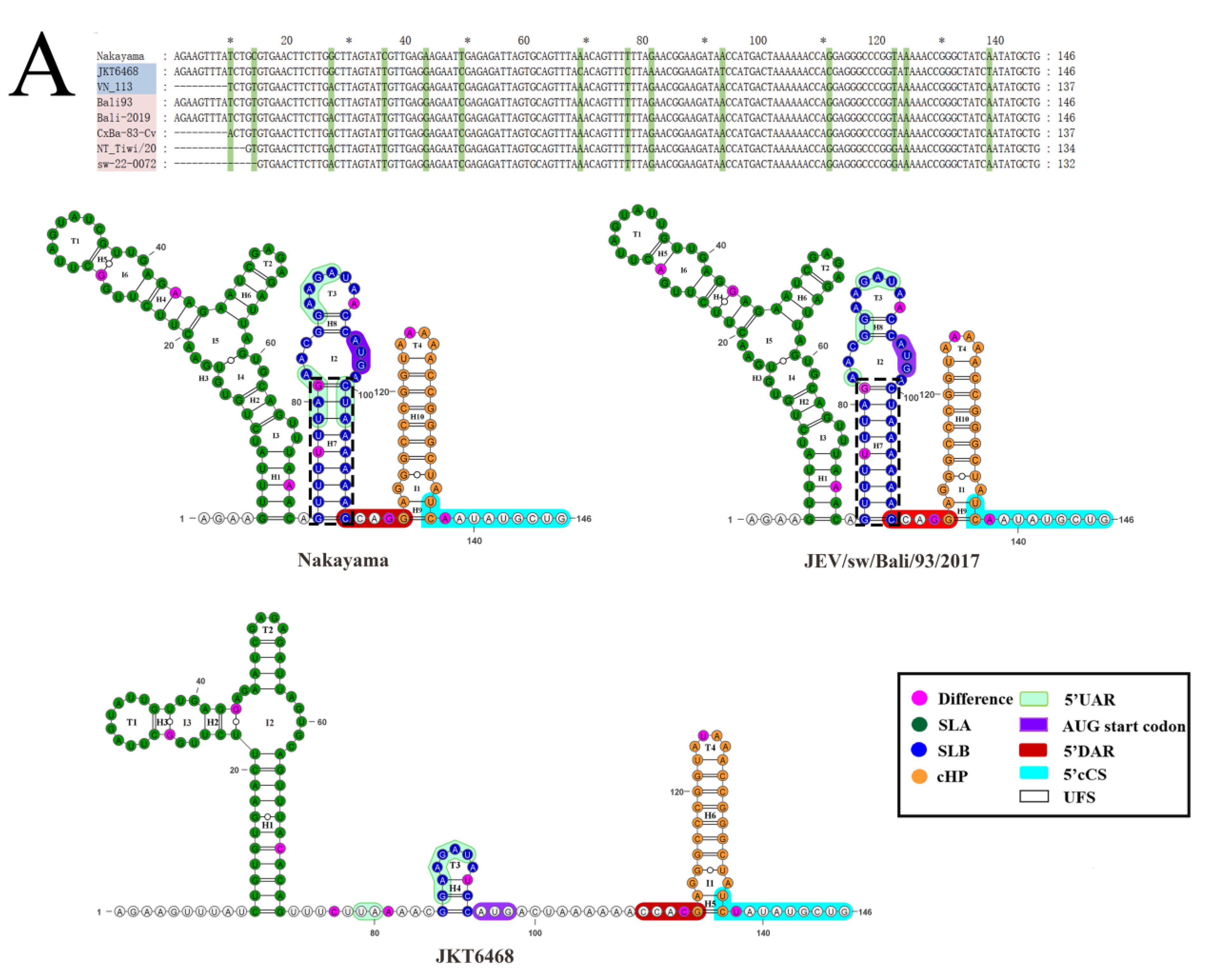
2.6.3. Comparison of the Sequence and Structure of the 5′ and 3′ UTRs of JEVs
2.6.4. Analysis of Amino Acid Mutations of Each Protein of GIV JEVs
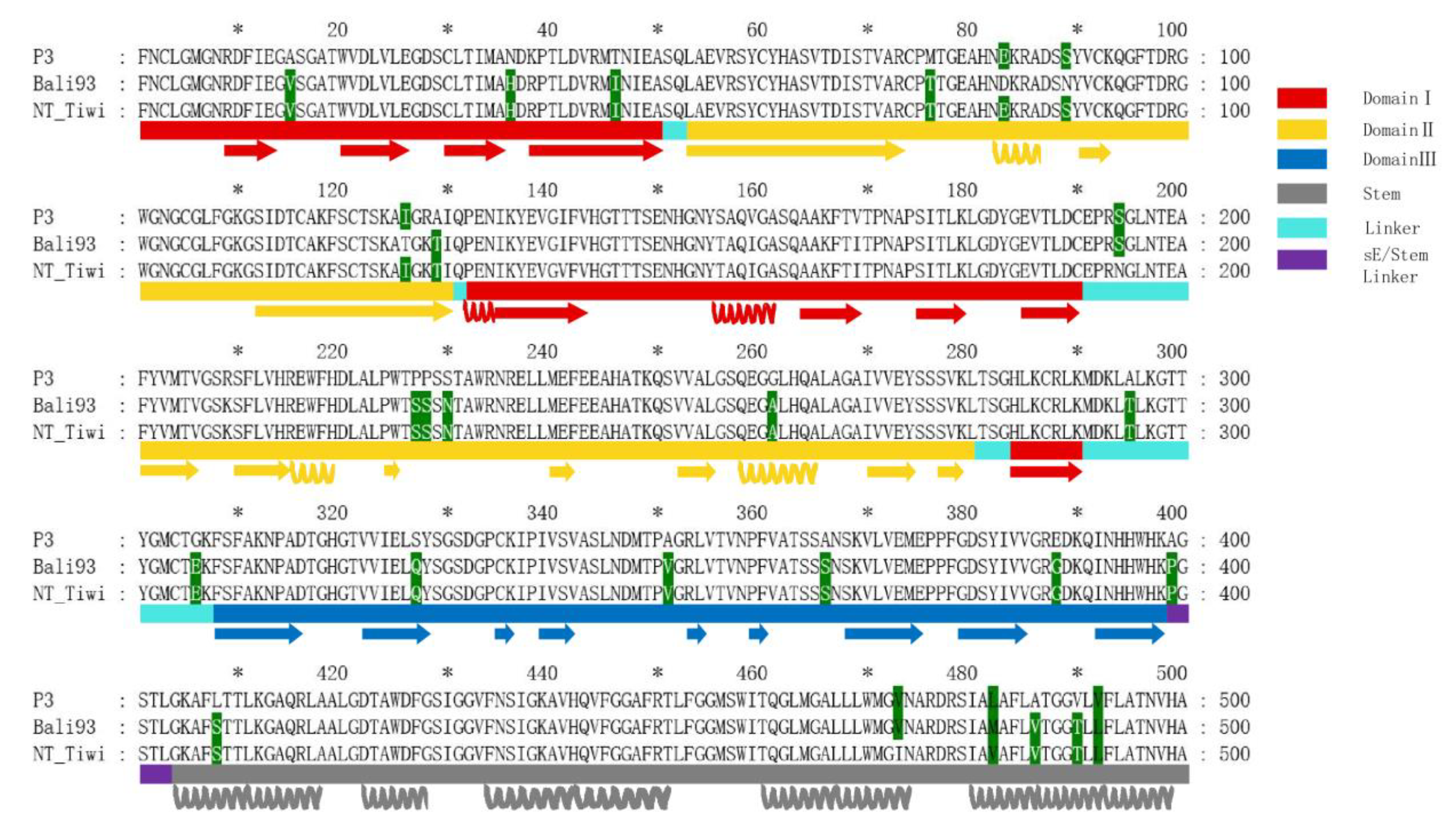
2.7. Comparative Structural Analysis of the Envelope Protein of Emerging GIV JEV Isolates and Vaccine Strain P3
3. Results
3.1. Complete Genome Sequence Dataset Construction
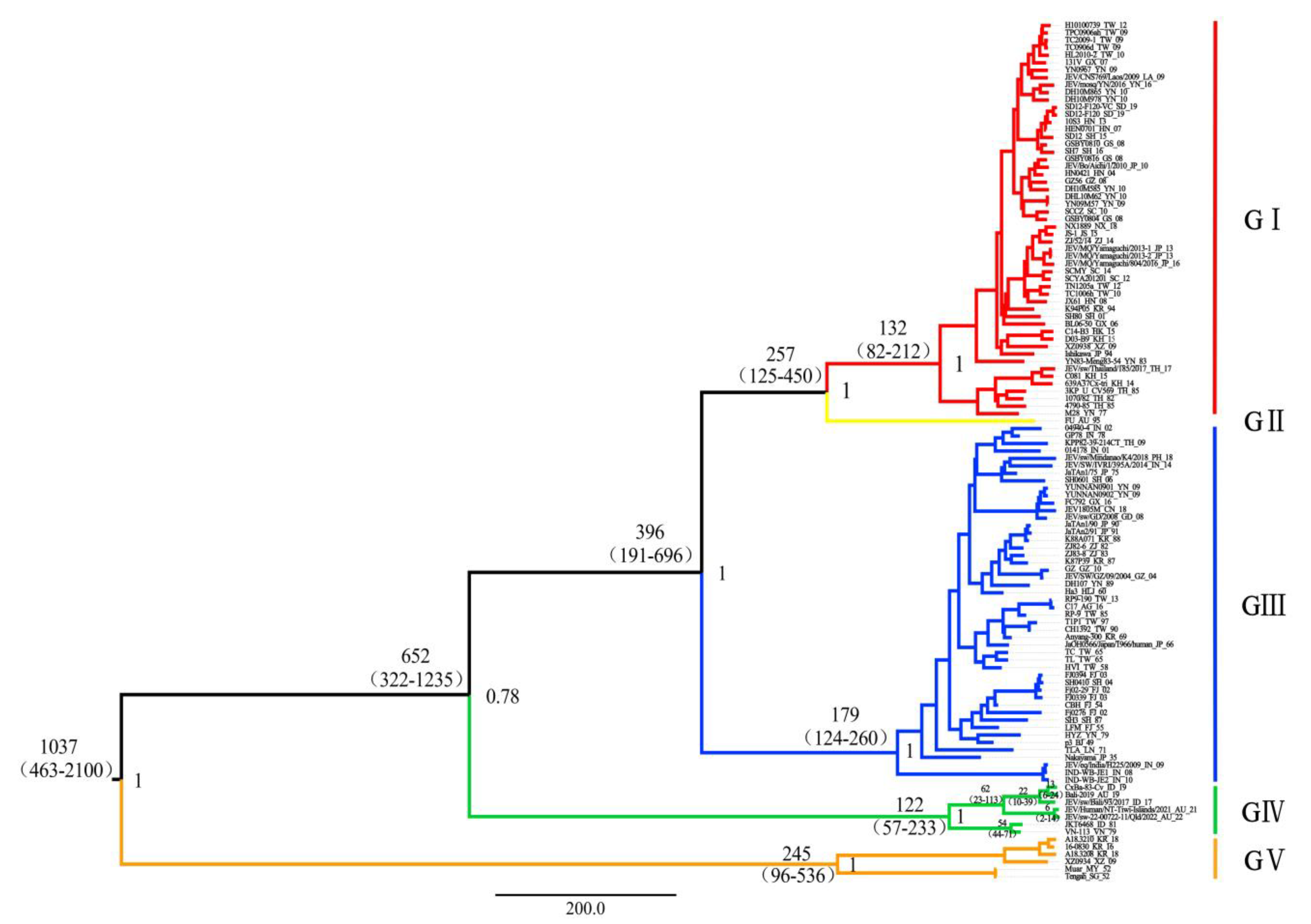
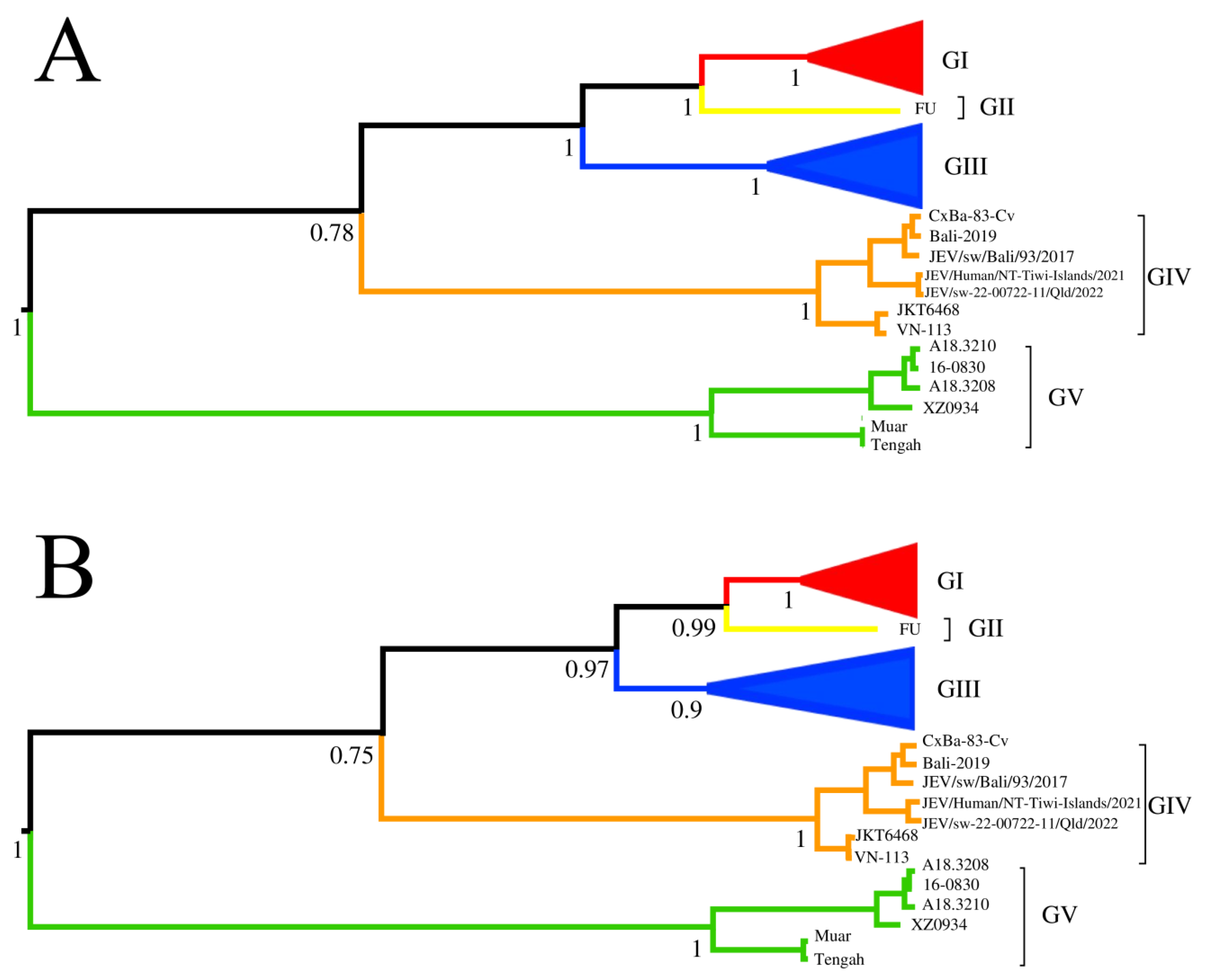
3.2. Time-Scaled Phylogenetic Evolutionary Analysis
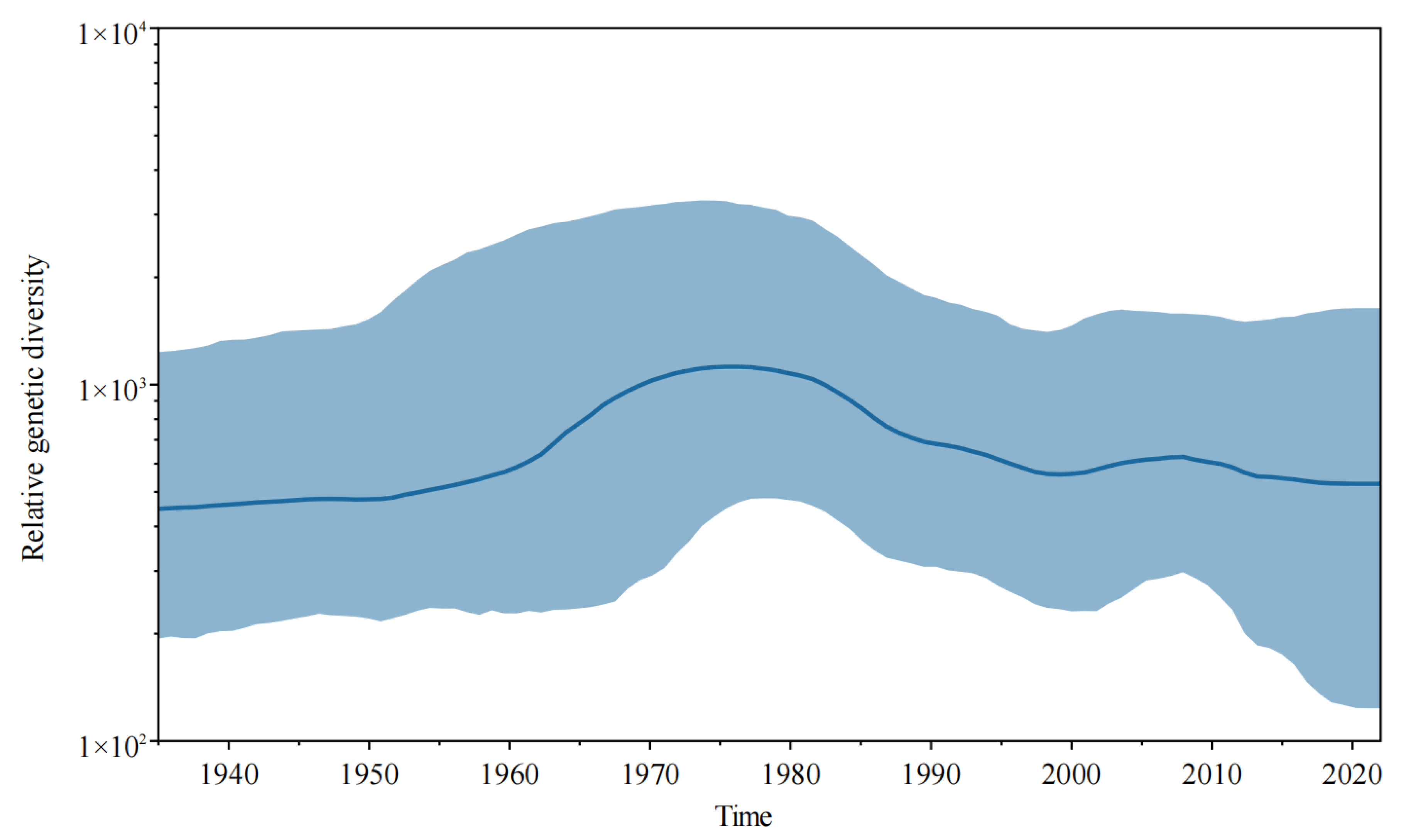
3.3. Evolutionary Rate and Population Dynamic Analysis
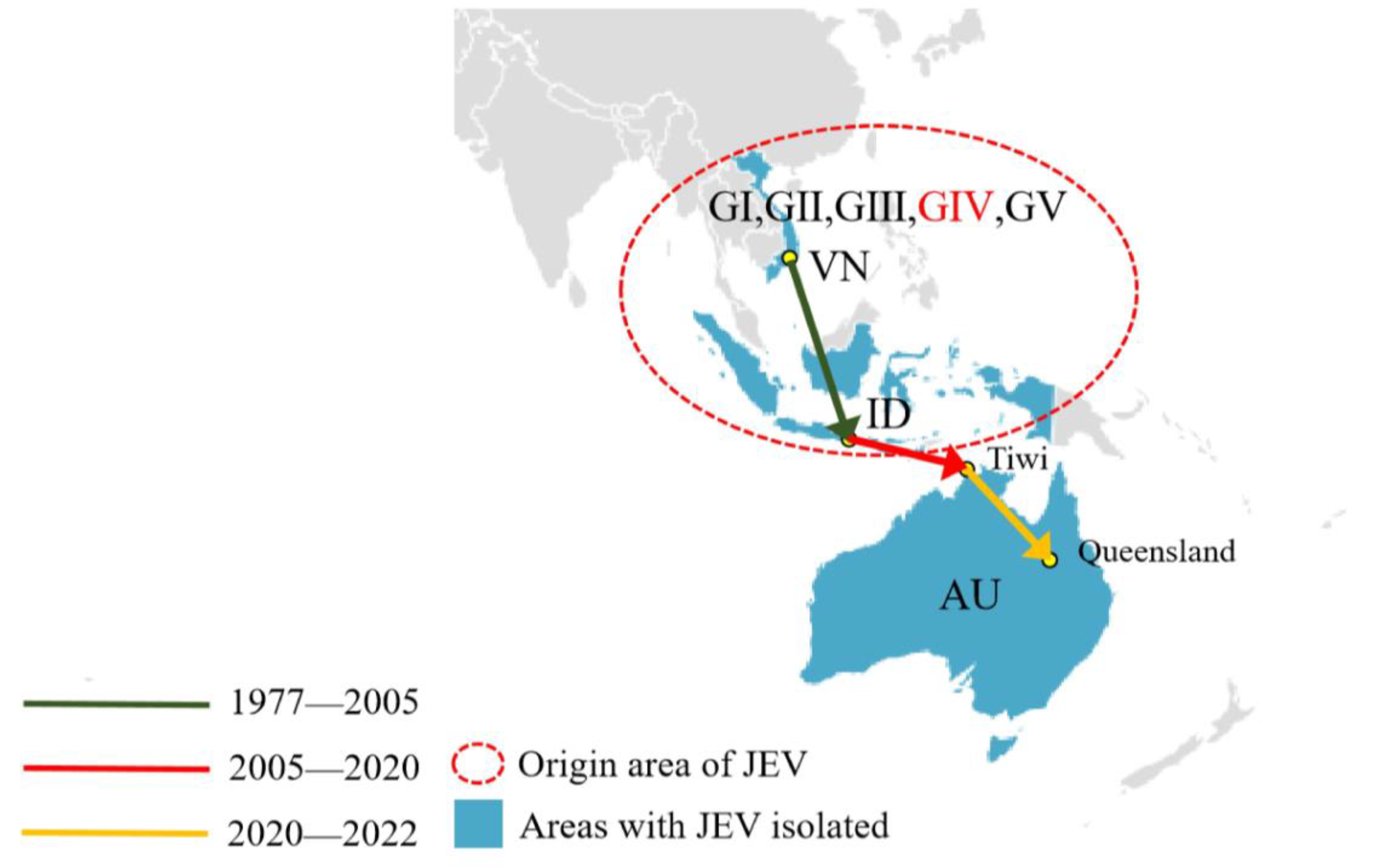
3.4. The Dispersal Route of JEV GIV Based on Phylogeographic Analysis
3.5. Phylogenetic Analysis of the Variable Region of JEVs
3.6. Similarity Analysis of the Nucleotides and Amino Acids of Different JEV Genotypes
3.7. Sequence Analysis of GIV JEVs
3.7.1. Similarity Analysis of the Whole Genome Sequence of GIV JEVs
3.7.2. Comparative Analysis of the Sequence and Structure of the UTRs of JEVs
3.7.3. Analysis of the Mutation Sites of Proteins within GIV JEVs
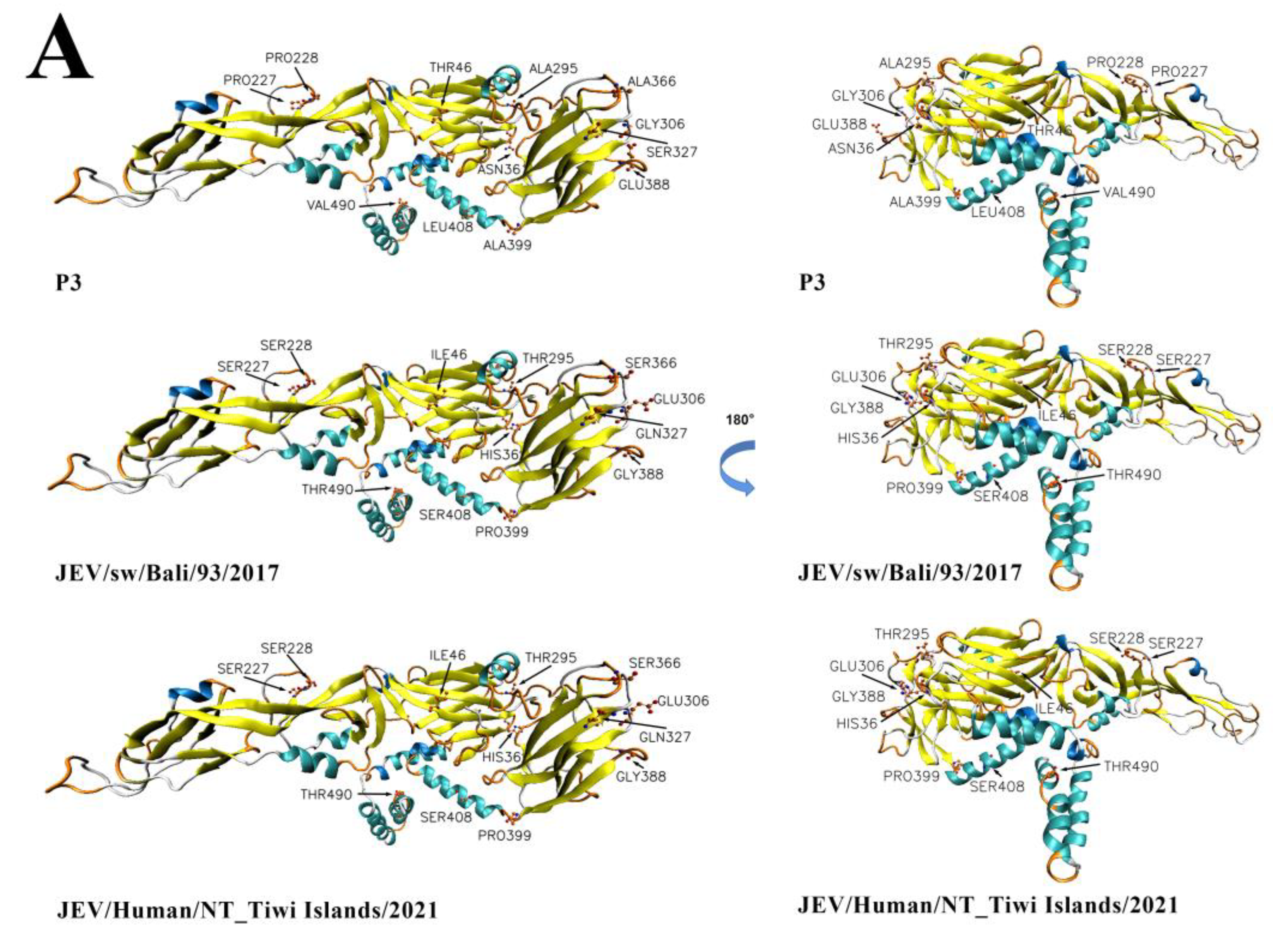
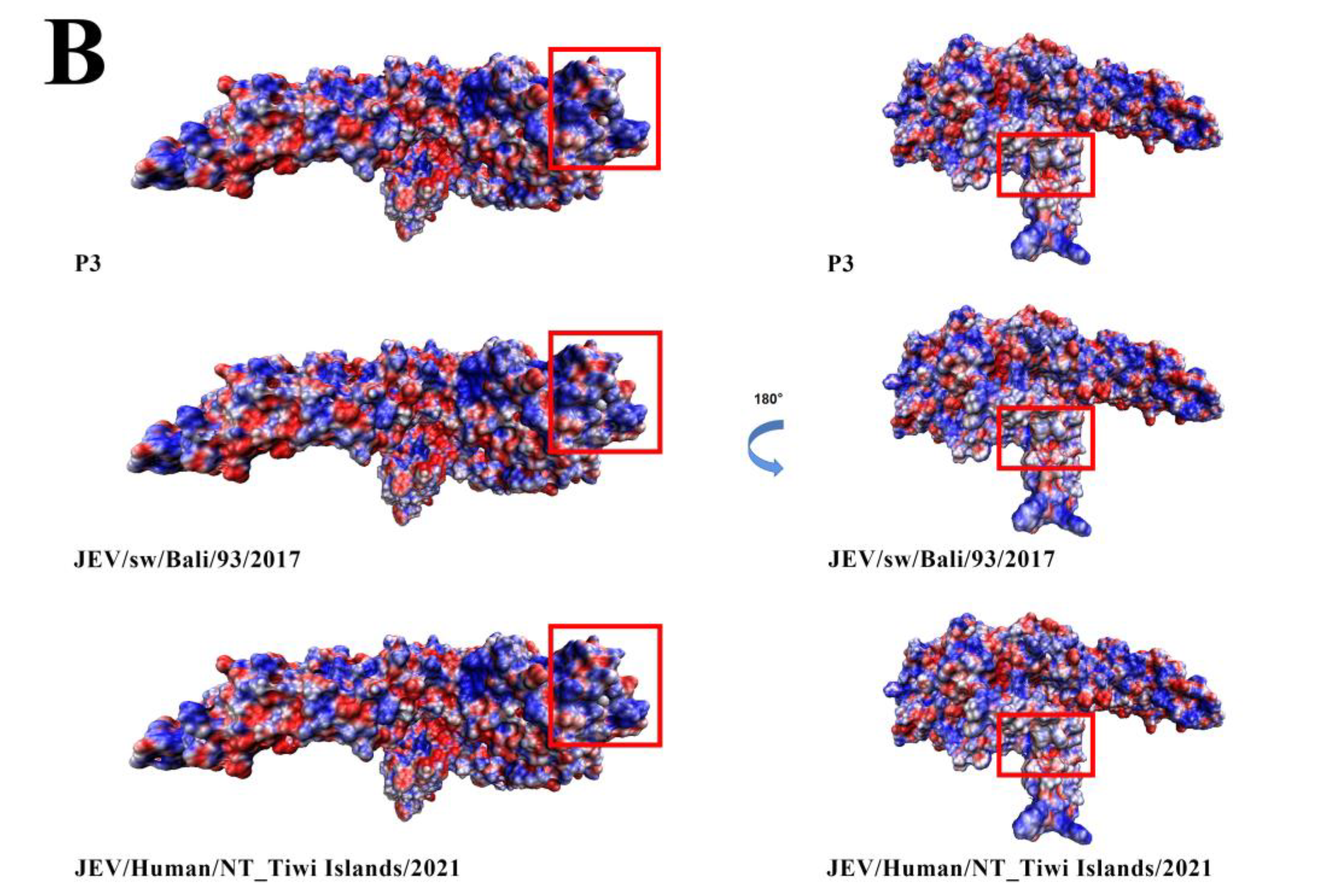
3.8. Comparative Analysis of the Envelope Protein between Emerging JEV GIV Isolates and Vaccine Strain P3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Halstead, S.B.; Jacobson, J. Japanese Encephalitis. In Advances in Virus Research; Chambers, T.J., Monath, T.P., Eds.; Academic Press: Cambridge, MA, USA, 2003; Volume 61, pp. 103–138. [Google Scholar]
- Campbell, G.L.; Hills, S.L.; Fischer, M.; Jacobson, J.A.; Hoke, C.H.; Hombach, J.M.; Marfin, A.A.; Solomon, T.; Tsai, T.F.; Tsu, V.D.; et al. Estimated global incidence of Japanese encephalitis: A systematic review. Bull. World Health Organ. 2011, 89, 766-774–774A–774E. [Google Scholar] [CrossRef]
- Lindenbach, B.; Thiel, H.J.; Rice, C.M. Flaviviridae: The viruses and their replication. Fields Virol. 2007, 1101–1151. [Google Scholar]
- Sumiyoshi, H.; Mori, C.; Fuke, I.; Morita, K.; Kuhara, S.; Kondou, J.; Kikuchi, Y.; Nagamatu, H.; Igarashi, A. Complete nucleotide sequence of the Japanese encephalitis virus genome RNA. Virology 1987, 161, 497–510. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J.; Niu, Y.; Liang, G. The 5’ and 3’ Untranslated Regions of the Japanese Encephalitis Virus (JEV): Molecular Genetics and Higher Order Structures. Front. Microbiol. 2021, 12, 730045. [Google Scholar] [CrossRef]
- Roy, U. Structural and molecular analyses of functional epitopes and escape mutants in Japanese encephalitis virus envelope protein domain III. Immunol. Res. 2020, 68, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Roy, U. Host–Virus Interactions in Japanese Encephalitis Virus. Zoonotic Dis. 2022, 2, 117–125. [Google Scholar] [CrossRef]
- Solomon, T.; Ni, H.; Beasley, D.W.; Ekkelenkamp, M.; Cardosa, M.J.; Barrett, A.D. Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 2003, 77, 3091–3098. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Liu, H.; Wang, H.; Fu, S.; Guo, Z.; Liang, G. Southernmost Asia is the source of Japanese encephalitis virus (genotype 1) diversity from which the viruses disperse and evolve throughout Asia. PLoS Negl. Trop. Dis. 2013, 7, e2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turtle, L.; Solomon, T. Japanese encephalitis—The prospects for new treatments. Nat. Rev. Neurol. 2018, 14, 298–313. [Google Scholar] [CrossRef]
- Pan, X.L.; Liu, H.; Wang, H.Y.; Fu, S.H.; Liu, H.Z.; Zhang, H.L.; Li, M.H.; Gao, X.Y.; Wang, J.L.; Sun, X.H.; et al. Emergence of genotype I of Japanese encephalitis virus as the dominant genotype in Asia. J. Virol. 2011, 85, 9847–9853. [Google Scholar] [CrossRef] [Green Version]
- Hanna, J.N.; Ritchie, S.A.; Phillips, D.A.; Shield, J.; Bailey, M.C.; Mackenzie, J.S.; Poidinger, M.; McCall, B.J.; Mills, P.J. An outbreak of Japanese encephalitis in the Torres Strait, Australia, 1995. Med. J. Aust. 1996, 165, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.T.; Wang, L.-F.; Daniels, P.W.; Mackenzie, J.S. Molecular characterization of the first Australian isolate of Japanese encephalitis virus, the FU strain. J. Gen. Virol. 2000, 81, 2471–2480. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-R.; Tesh, R.B.; Rico-Hesse, R. Genetic Variation of Japanese Encephalitis Virus in Nature. J. Gen. Virol. 1990, 71, 2915–2922. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Fu, S.H.; Chen, W.X.; Wang, H.Y.; Guo, Y.H.; Liu, Q.Y.; Li, Y.X.; Luo, H.M.; Da, W.; Duo Ji, D.Z.; et al. Genotype v Japanese encephalitis virus is emerging. PLoS Negl. Trop. Dis. 2011, 5, e1231. [Google Scholar] [CrossRef] [PubMed]
- Li, M.H.; Fu, S.H.; Chen, W.X.; Wang, H.Y.; Cao, Y.X.; Liang, G.D. Molecular characterization of full-length genome of Japanese encephalitis virus genotype V isolated from Tibet, China. Biomed. Environ. Sci. 2014, 27, 231–239. [Google Scholar] [PubMed]
- Takhampunya, R.; Kim, H.-C.; Tippayachai, B.; Kengluecha, A.; Klein, T.A.; Lee, W.-J.; Grieco, J.; Evans, B.P. Emergence of Japanese encephalitis virus genotype V in the Republic of Korea. Virol. J. 2011, 8, 449. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Cha, G.W.; Jeong, Y.E.; Lee, W.G.; Chang, K.S.; Roh, J.Y.; Yang, S.C.; Park, M.Y.; Park, C.; Shin, E.H. Detection of Japanese encephalitis virus genotype V in Culex orientalis and Culex pipiens (Diptera: Culicidae) in Korea. PLoS ONE 2015, 10, e0116547. [Google Scholar] [CrossRef] [Green Version]
- Woo, J.H.; Jeong, Y.E.; Jo, J.E.; Shim, S.M.; Ryou, J.; Kim, K.C.; Lee, W.J.; Lee, J.Y. Genetic Characterization of Japanese Encephalitis Virus Genotype 5 Isolated from Patient, South Korea, 2015. Emerg. Infect. Dis. 2020, 26, 1002–1006. [Google Scholar] [CrossRef]
- Kim, H.C.; Takhampunya, R.; Tippayachai, B.; Chong, S.T.; Park, J.Y.; Kim, M.S.; Seo, H.J.; Yeh, J.Y.; Lee, W.J.; Lee, D.K.; et al. Japanese encephalitis virus in culicine mosquitoes (Diptera: Culicidae) of the republic of Korea, 2008–2010. Mil. Med. 2015, 180, 158–167. [Google Scholar] [CrossRef] [Green Version]
- Sanborn, M.A.; Wuertz, K.M.; Kim, H.C.; Yang, Y.; Li, T.; Pollett, S.D.; Jarman, R.G.; Berry, I.M.; Klein, T.A.; Hang, J. Metagenomic analysis reveals Culex mosquito virome diversity and Japanese encephalitis genotype V in the Republic of Korea. Mol. Ecol. 2021, 30, 5470–5487. [Google Scholar] [CrossRef]
- Mackenzie, J.S.; Johansen, C.A.; Ritchie, S.A.; van den Hurk, A.F.; Hall, R.A. Japanese Encephalitis as an Emerging Virus: The Emergence and Spread of Japanese Encephalitis Virus in Australasia. In Japanese Encephalitis and West Nile Viruses; Mackenzie, J.S., Barrett, A.D.T., Deubel, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2002; pp. 49–73. [Google Scholar]
- Kuwata, R.; Torii, S.; Shimoda, H.; Supriyono, S.; Phichitraslip, T.; Prasertsincharoen, N.; Takemae, H.; Bautista, R.; Ebora, V.; Abella, J.A.C.; et al. Distribution of Japanese Encephalitis Virus, Japan and Southeast Asia, 2016–2018. Emerg. Infect. Dis. 2020, 26, 125–128. [Google Scholar] [CrossRef] [Green Version]
- Faizah, A.N.; Kobayashi, D.; Maekawa, Y.; Amoa-Bosompem, M.; Fauziyah, S.; Mulyatno, K.C.; Subekti, S.; Rohmah, E.A.; Lusida, M.I.; Mori, Y.; et al. Identification and Isolation of Japanese Encephalitis Virus Genotype IV from Culex vishnui Collected in Bali, Indonesia in 2019. Am. J. Trop. Med. Hyg. 2021, 105, 813–817. [Google Scholar] [CrossRef]
- Pyke, A.T.; Choong, K.; Moore, F.; Schlebusch, S.; Taylor, C.; Hewitson, G.; McMahon, J.; Nair, N.; Moore, P.; Finger, M.; et al. A Case of Japanese Encephalitis with a Fatal Outcome in an Australian Who Traveled from Bali in 2019. Trop. Med. Infect. Dis. 2020, 5, 133. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.S.; Williams, D.T.; van den Hurk, A.F.; Smith, D.W.; Currie, B.J. Japanese Encephalitis Virus: The Emergence of Genotype IV in Australia and Its Potential Endemicity. Viruses 2022, 14, 2480. [Google Scholar] [CrossRef] [PubMed]
- Sikazwe, C.; Neave, M.J.; Michie, A.; Mileto, P.; Wang, J.; Cooper, N.; Levy, A.; Imrie, A.; Baird, R.W.; Currie, B.J.; et al. Molecular detection and characterisation of the first Japanese encephalitis virus belonging to genotype IV acquired in Australia. PLoS Negl. Trop. Dis. 2022, 16, e0010754. [Google Scholar] [CrossRef] [PubMed]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Lemey, P.; Lott, M.; Moulton, V.; Posada, D.; Lefeuvre, P. RDP3: A flexible and fast computer program for analyzing recombination. Bioinformatics 2010, 26, 2462–2463. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lole Kavita, S.; Bollinger Robert, C.; Paranjape Ramesh, S.; Gadkari, D.; Kulkarni Smita, S.; Novak Nicole, G.; Ingersoll, R.; Sheppard Haynes, W.; Ray Stuart, C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Land, H.; Humble, M.S. YASARA: A Tool to Obtain Structural Guidance in Biocatalytic Investigations. Methods Mol. Biol. 2018, 1685, 43–67. [Google Scholar] [PubMed]
- Gao, X.; Liu, H.; Li, M.; Fu, S.; Liang, G. Insights into the evolutionary history of Japanese encephalitis virus (JEV) based on whole-genome sequences comprising the five genotypes. Virol. J. 2015, 12, 43. [Google Scholar] [CrossRef] [Green Version]
- Hussain-Alkhateeb, L.; Rivera Ramírez, T.; Kroeger, A.; Gozzer, E.; Runge-Ranzinger, S. Early warning systems (EWSs) for chikungunya, dengue, malaria, yellow fever, and Zika outbreaks: What is the evidence? A scoping review. PLoS Negl. Trop. Dis. 2021, 15, e0009686. [Google Scholar] [CrossRef]
- Uzcategui, N.Y.; Sironen, T.; Golovljova, I.; Jaaskelainen, A.E.; Valimaa, H.; Lundkvist, A.; Plyusnin, A.; Vaheri, A.; Vapalahti, O. Rate of evolution and molecular epidemiology of tick-borne encephalitis virus in Europe, including two isolations from the same focus 44 years apart. J. Gen. Virol. 2012, 93, 786–796. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of West Nile virus: From the cradle of evolution in Africa to Eurasia, Australia, and the Americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef] [Green Version]
- Bryant, J.E.; Holmes, E.C.; Barrett, A.D. Out of Africa: A molecular perspective on the introduction of yellow fever virus into the Americas. PLoS Pathog. 2007, 3, e75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalev, S.Y.; Mukhacheva, T.A. Tick-borne encephalitis virus subtypes emerged through rapid vector switches rather than gradual evolution. Ecol. Evol. 2014, 4, 4307–4316. [Google Scholar] [CrossRef]
- Liu, H.; Shen, L.; Zhang, X.L.; Li, X.L.; Liang, G.D.; Ji, H.F. From discovery to outbreak: The genetic evolution of the emerging Zika virus. Emerg. Microbes Infect. 2016, 5, e111. [Google Scholar] [CrossRef] [Green Version]
- Muniraju, M.; Munir, M.; Parthiban, A.R.; Banyard, A.C.; Bao, J.; Wang, Z.; Ayebazibwe, C.; Ayelet, G.; El Harrak, M.; Mahapatra, M.; et al. Molecular evolution of peste des petits ruminants virus. Emerg. Infect. Dis. 2014, 20, 2023–2033. [Google Scholar] [CrossRef] [Green Version]
- Anez, G.; Grinev, A.; Chancey, C.; Ball, C.; Akolkar, N.; Land, K.J.; Winkelman, V.; Stramer, S.L.; Kramer, L.D.; Rios, M. Evolutionary dynamics of West Nile virus in the United States, 1999–2011: Phylogeny, selection pressure and evolutionary time-scale analysis. PLoS Negl. Trop. Dis. 2013, 7, e2245. [Google Scholar] [CrossRef] [Green Version]
- Egyed, L.; Ronai, Z.; Dan, A. Hungarian tick-borne encephalitis viruses isolated from a 0.5-ha focus are closely related to Finnish strains. Ticks Tick Borne Dis. 2018, 9, 1064–1068. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popović, M.; Poljšak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodušek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016, 374, 951–958. [Google Scholar] [CrossRef]
- Hale, J.H.; Lim, K.A.; Chee, P.H. Japanese type B encephalitis in Malaya. Ann. Trop. Med. Parasitol. 1952, 46, 220–226. [Google Scholar] [CrossRef]
- Hasegawa, H.; Yoshida, M.; Fujita, S.; Kobayashi, Y. Comparison of structural proteins among antigenically different Japanese encephalitis virus strains. Vaccine 1994, 12, 841–844. [Google Scholar] [CrossRef]
- Ritchie, S.A.; Rochester, W. Wind-blown mosquitoes and introduction of Japanese encephalitis into Australia. Emerg. Infect. Dis. 2001, 7, 900–903. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.; Chang, G.; Yu, Y. Japanese Encephalitis Vaccines, 3rd ed.; Plotkin, S.A., Orenstein, W.A., Eds.; WB Saunders: Philadelphia, PA, USA, 1999; pp. 672–703. [Google Scholar]
- Erra, E.O.; Askling, H.H.; Yoksan, S.; Rombo, L.; Riutta, J.; Vene, S.; Lindquist, L.; Vapalahti, O.; Kantele, A. Cross-protective capacity of Japanese encephalitis (JE) vaccines against circulating heterologous JE virus genotypes. Clin. Infect. Dis. 2013, 56, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Fu, S.; Gao, X.; Li, M.; Cui, S.; Li, X.; Cao, Y.; Lei, W.; Lu, Z.; He, Y.; et al. Low Protective Efficacy of the Current Japanese Encephalitis Vaccine against the Emerging Genotype 5 Japanese Encephalitis Virus. PLoS Negl. Trop. Dis. 2016, 10, e0004686. [Google Scholar] [CrossRef] [PubMed] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO | Strains | Date | Locations | Genotype | Locus |
|---|---|---|---|---|---|
| 1 | M28 | 1977 | China | I | JF706279.1 |
| 2 | 1070/82 (Subin) | 1982 | Thailand | I | GQ902059.1 |
| 3 | YN83-Meng83–54 | 1983 | China: Yunnan | I | JF706282.1 |
| 4 | 3KP’’U’’CV569 | 1985 | Thailand | I | GQ902060.1 |
| 5 | 4790-85 | 1985 | Thailand | I | GQ902062.1 |
| 6 | Ishikawa | 1994 | Japan | I | AB051292.1 |
| 7 | K94P05 | 1994 | Korea | I | AF045551.2 |
| 8 | SH-80 | 2001 | China: Shanghai | I | JN381848.1 |
| 9 | HN0421 | 2004 | China: Henan | I | JN381841.1 |
| 10 | BL06-50 | 2006 | China: Guangxi | I | JF706270.1 |
| 11 | 131V | 2007 | China: Guangxi | I | GU205163.1 |
| 12 | HEN0701 | 2007 | China: Henan | I | FJ495189.1 |
| 13 | JX61 | 2008 | China: Henan | I | GU556217.1 |
| 14 | GSBY0816 | 2008 | China: Gansu | I | JN381842.1 |
| 15 | GSBY0804 | 2008 | China: Gansu | I | JN381844.1 |
| 16 | GSBY0810 | 2008 | China: Gansu | I | JN381840.1 |
| 17 | GZ56 | 2008 | China: Guizhou | I | HM366552.1 |
| 18 | YN09M57 | 2009 | China: Yunnan | I | KT229574.1 |
| 19 | JEV/CNS769/Laos/2009 | 2009 | Laos | I | KC196115.1 |
| 20 | YN0967 | 2009 | China: Yunnan | I | JF706268.1 |
| 21 | JEV/Taiwan/TC0906d/M/2009 | 2009 | China: Taiwan | I | KF667320.1 |
| 22 | JEV/Taiwan/TPC0906ah/M/2009 | 2009 | China: Taiwan | I | KF667318.1 |
| 23 | XZ0938 | 2009 | China: Xizang | I | HQ652538.1 |
| 24 | TC2009-1 | 2009 | China: Taiwan | I | JF499790.1 |
| 25 | DH10M978 | 2010 | China: Yunnan | I | KT229573.1 |
| 26 | DH10M865 | 2010 | China: Yunnan | I | KT229572.1 |
| 27 | DHL10M62 | 2010 | China: Yunnan | I | KT229575.1 |
| 28 | SCCZ | 2010 | China: Sichuan | I | KU351667.1 |
| 29 | HL2010-2 | 2010 | China: Taiwan | I | JQ031753.1 |
| 30 | JEV/Bo/Aichi/1/2010 | 2010 | Japan: Aichi | I | AB853904.1 |
| 31 | JEV/Taiwan/TC1006h/M/2010 | 2010 | China: Taiwan | I | KF667321.1 |
| 32 | DH10M585 | 2010 | China: Yunnan | I | KT957421.1 |
| 33 | SCYA201201 | 2012 | China: Sichuan | I | KM658163.1 |
| 34 | JEV/Taiwan/TN1205a/M/2012(2) | 2012 | China: Taiwan | I | KF667325.1 |
| 35 | JEV/Taiwan/H10100739/H/2012 | 2012 | China: Taiwan | I | KF667324.1 |
| 36 | JEV/MQ/Yamaguchi/2013/2 | 2013 | Japan: Yamaguchi, Yoshida | I | AB981184.1 |
| 37 | JEV/MQ/Yamaguchi/2013/1 | 2013 | Japan: Yamaguchi, Yoshida | I | AB981183.1 |
| 38 | 10S3 | 2013 | China: Henan | I | MF542268.1 |
| 39 | SCMY | 2014 | China: Sichuan | I | KU351668.1 |
| 40 | ZJ/52/14 | 2014 | China | I | MK558811.1 |
| 41 | 639A37Cx-tri | 2014 | Cambodia | I | KY927815.1 |
| 42 | JS-1 | 2015 | China: Jiangsu | I | KX357114.1 |
| 43 | C081 | 2015 | Cambodia | I | KY927816.1 |
| 44 | C14-B3 | 2015 | Cambodia | I | KY927817.1 |
| 45 | D03-B9 | 2015 | Cambodia | I | KY927818.1 |
| 46 | SD12 | 2015 | China: Shanghai | I | MH753127.1 |
| 47 | JEV/MQ/Yamaguchi/804/2016 | 2016 | Japan: Yamaguchi, Yoshida | I | LC461957.1 |
| 48 | JEV/mosq/YN/2016 | 2016 | China | I | MH385014.1 |
| 49 | SH7 | 2016 | China: Shanghai | I | MH753129.1 |
| 50 | JEV/sw/Thailand/185/2017 | 2017 | Thailand | I | LC461958.1 |
| 51 | NX1889 | 2018 | China: Ningxia | I | MT134112.1 |
| 52 | SD12-F120 | 2019 | China | I | MN544779.1 |
| 53 | SD12-F120-VC | 2019 | China | I | MN544780.1 |
| 54 | FU | 1995 | Australia | II | AF217620.1 |
| 55 | Nakayama | 1935 | Japan | III | EF571853.1 |
| 56 | p3 | 1949 | China: Beijing | III | U47032.1 |
| 57 | CBH | 1954 | China: Fujian | III | JN381860.1 |
| 58 | LFM | 1955 | China: Fujian | III | JN381863.1 |
| 59 | HVI | 1965 | China: Taiwan | III | AF098735.1 |
| 60 | Ha3 | 1960′s | China: Heilongjiang | III | JN381872.1 |
| 61 | TL | 1965 | China: Taiwan | III | AF098737.1 |
| 62 | TC | 1965 | China: Taiwan | III | AF098736.1 |
| 63 | JaOH0566/Japan/1966/human | 1966 | Japan | III | AY508813.1 |
| 64 | Anyang-300 | 1969 | South Korea | III | KT447437.1 |
| 65 | TLA | 1971 | China: Liaoning | III | JN381868.1 |
| 66 | JaTAn1/75 | 1975 | Japan: Tokyo | III | AB551990.1 |
| 67 | GP78 | 1978 | India | III | AF075723.1 |
| 68 | HYZ | 1979 | China: Yunnan | III | JN381853.1 |
| 69 | ZJ82-6 | 1982 | China: Zhejiang | III | KY650724.1 |
| 70 | ZJ83-8 | 1983 | China: Zhejiang | III | KY650725.1 |
| 71 | RP-9 | 1985 | China: Taiwan | III | AF014161.1 |
| 72 | SH3 | 1987 | China: Shanghai | III | JN381864.1 |
| 73 | K87P39 | 1987 | South Korea | III | AY585242.1 |
| 74 | K88A071 | 1988 | South Korea | III | KR908703.1 |
| 75 | DH107 | 1989 | China: Yunnan | III | JN381873.1 |
| 76 | CH1392 | 1990 | China: Taiwan | III | AF254452.1 |
| 77 | JaTAn1/90 | 1990 | Japan: Tokyo | III | AB551991.1 |
| 78 | JaTAn2/91 | 1991 | Japan: Tokyo | III | AB551992.1 |
| 79 | T1P1 | 1997 | China: Taiwan: Liu-Chiu islet | III | AF254453.1 |
| 80 | 14178 | 2001 | India | III | EF623987.1 |
| 81 | Fj02-29 | 2002 | China: Fujian | III | JF706273.1 |
| 82 | 04940-4 | 2002 | India: Maharashtra, Bhandara district | III | EF623989.1 |
| 83 | Fj0276 | 2002 | China: Fujian | III | JN381867.1 |
| 84 | FJ0339 | 2003 | China: Fujian | III | JN381859.1 |
| 85 | FJ0394 | 2003 | China: Fujian | III | JN381858.1 |
| 86 | SH0410 | 2004 | China: Shanghai | III | JN381856.1 |
| 87 | JEV/SW/GZ/09/2004 | 2004 | China: Guizhou | III | KF297916.1 |
| 88 | SH0601 | 2006 | China: Shanghai | III | EF543861.1 |
| 89 | IND-WB-JE1 | 2008 | India: West Bengal | III | JX050179.1 |
| 90 | JEV/sw/GD/2008 | 2008 | China: Guangdong | III | KX965684.1 |
| 91 | KPP82-39-214CT | 2009 | Thailand | III | GQ902063.1 |
| 92 | JEV/eq/India/H225/2009 | 2009 | India | III | JX131374.1 |
| 93 | YUNNAN0902 | 2009 | China: Yunnan Province | III | JQ086763.1 |
| 94 | YUNNAN0901 | 2009 | China: Yunnan | III | JQ086762.1 |
| 95 | IND-WB-JE2 | 2010 | India: West Bengal | III | JX072965.1 |
| 96 | GZ | 2010 | China: Guizhou | III | KC915016.1 |
| 97 | RP9-190 | 2013 | China: Taiwan | III | KF907505.1 |
| 98 | JEV/SW/IVRI/395A/2014 | 2014 | India | III | KP164498.2 |
| 99 | C17 | 2016 | Angola | III | KX945367.1 |
| 100 | FC792 | 2016 | China: Guangxi | III | MF002373.1 |
| 101 | JEV1805M | 2018 | China | III | MN639770.1 |
| 102 | JEV/sw/Mindanao/K4/2018 | 2018 | Philippines: Mindanao | III | LC461960.1 |
| 103 | VN 113 | 1979 | Viet Nam | IV | KU705228.1 |
| 104 | JKT6468 | 1981 | Indonesia | IV | AY184212.1 |
| 105 | JEV/sw/Bali/93/2017 | 2017 | Indonesia: Bali, Denpasar | IV | LC461961.1 |
| 106 | Bali 2019 | 2019 | Australia | IV | MT253731.1 |
| 107 | 19CxBa-83-Cv | 2019 | Indonesia: Bali | IV | LC579814.1 |
| 108 | JEV/Human/NT_Tiwi Islands/2021 | 2021 | Australia: Northern Territory, Tiwi Islands | IV | OM867669.1 |
| 109 | JEV/sw-22-00722-11/Qld/2022 | 2022 | Australia: Queensland | IV | ON624132.1 |
| 110 | Muar | 1952 | Malaysia | V | HM596272.1 |
| 111 | Tengah | 1952 | Singapore | V | KM677246.1 |
| 112 | XZ0934 | 2009 | China: Xizang | V | JF915894.1 |
| 113 | A18.3210 | 2018 | South Korea: Camp Humphreys | V | MT568538.1 |
| 114 | A18.3208 | 2018 | South Korea: Camp Humphreys | V | MT568539.1 |
| 115 | 16-0830 | 2016 | South Korea: Yongsan | V | MT568540.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, G.; Gao, T.; Wang, Z.; Zhang, J.; Cui, B.; Shen, X.; Zhou, A.; Zhang, Y.; Zhao, J.; Liu, H.; et al. Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution. Viruses 2023, 15, 626. https://doi.org/10.3390/v15030626
Xu G, Gao T, Wang Z, Zhang J, Cui B, Shen X, Zhou A, Zhang Y, Zhao J, Liu H, et al. Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution. Viruses. 2023; 15(3):626. https://doi.org/10.3390/v15030626
Chicago/Turabian StyleXu, Guanlun, Tingting Gao, Zhijie Wang, Jun Zhang, Baoqiu Cui, Xinxin Shen, Anyang Zhou, Yuan Zhang, Jie Zhao, Hong Liu, and et al. 2023. "Re-Emerged Genotype IV of Japanese Encephalitis Virus Is the Youngest Virus in Evolution" Viruses 15, no. 3: 626. https://doi.org/10.3390/v15030626