
Whole-Genome Analysis of Influenza A(H3N2) and B/Victoria Viruses Detected in Myanmar during the COVID-19 Pandemic in 2021

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Laboratory Confirmation
2.2.1. Multiplex PCR Screening
2.2.2. RNA Extraction
2.2.3. Cycling Probe Real-Time PCR for Subtyping, Lineage Detection and Ct Value Evaluation
2.2.4. Genetic Analysis by Next-Generation Sequencing
2.2.5. NGS Data Analysis
2.3. Phylogenetic Analysis
2.4. Ethical Considerations
3. Results
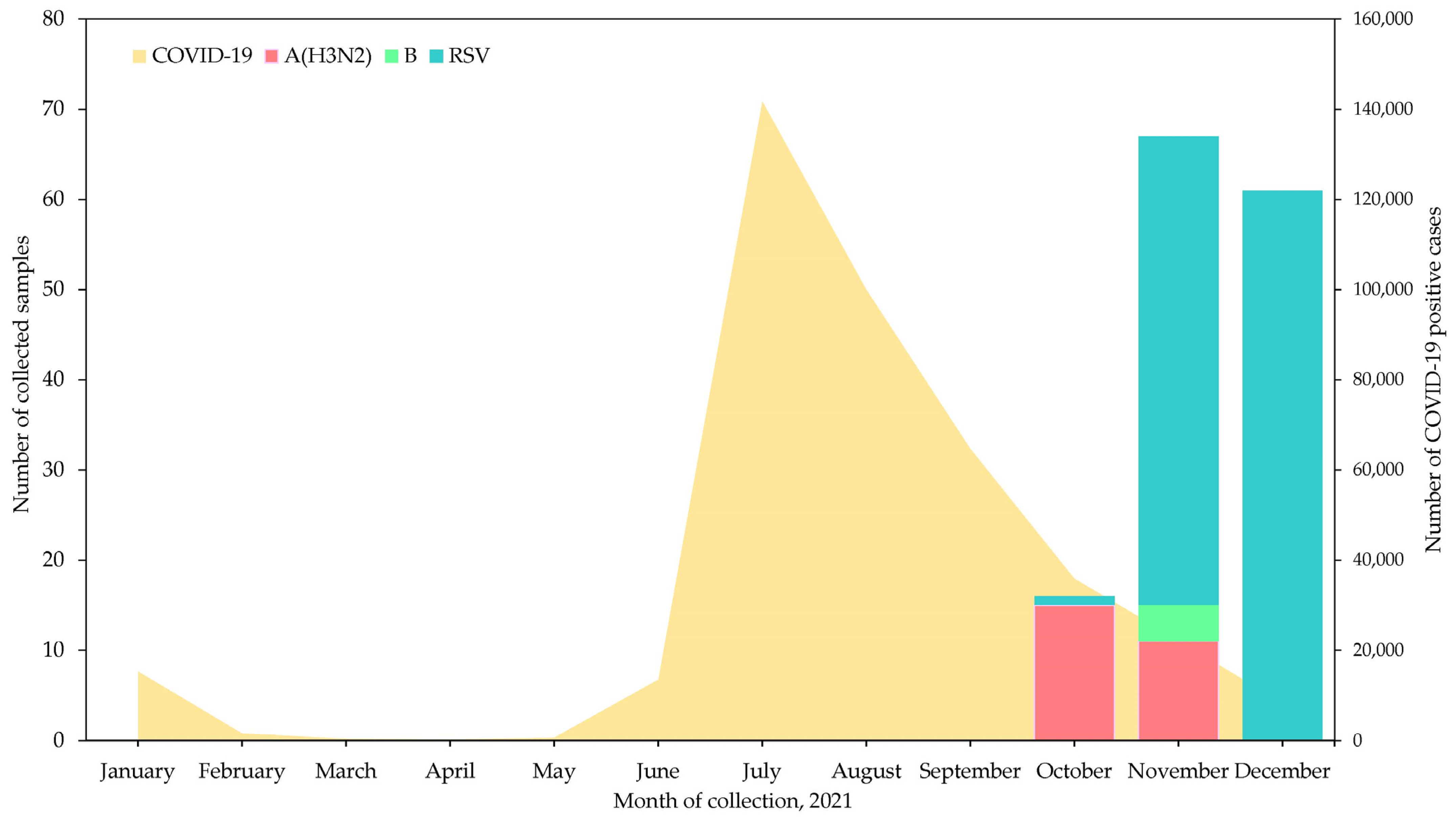
3.1. Number of Samples and Detection of Influenza Virus Types and Subtypes
3.2. Clinical Characteristics of Patients in This Study
3.3. Next-Generation Sequencing of Influenza Viruses Collected in Myanmar in 2021
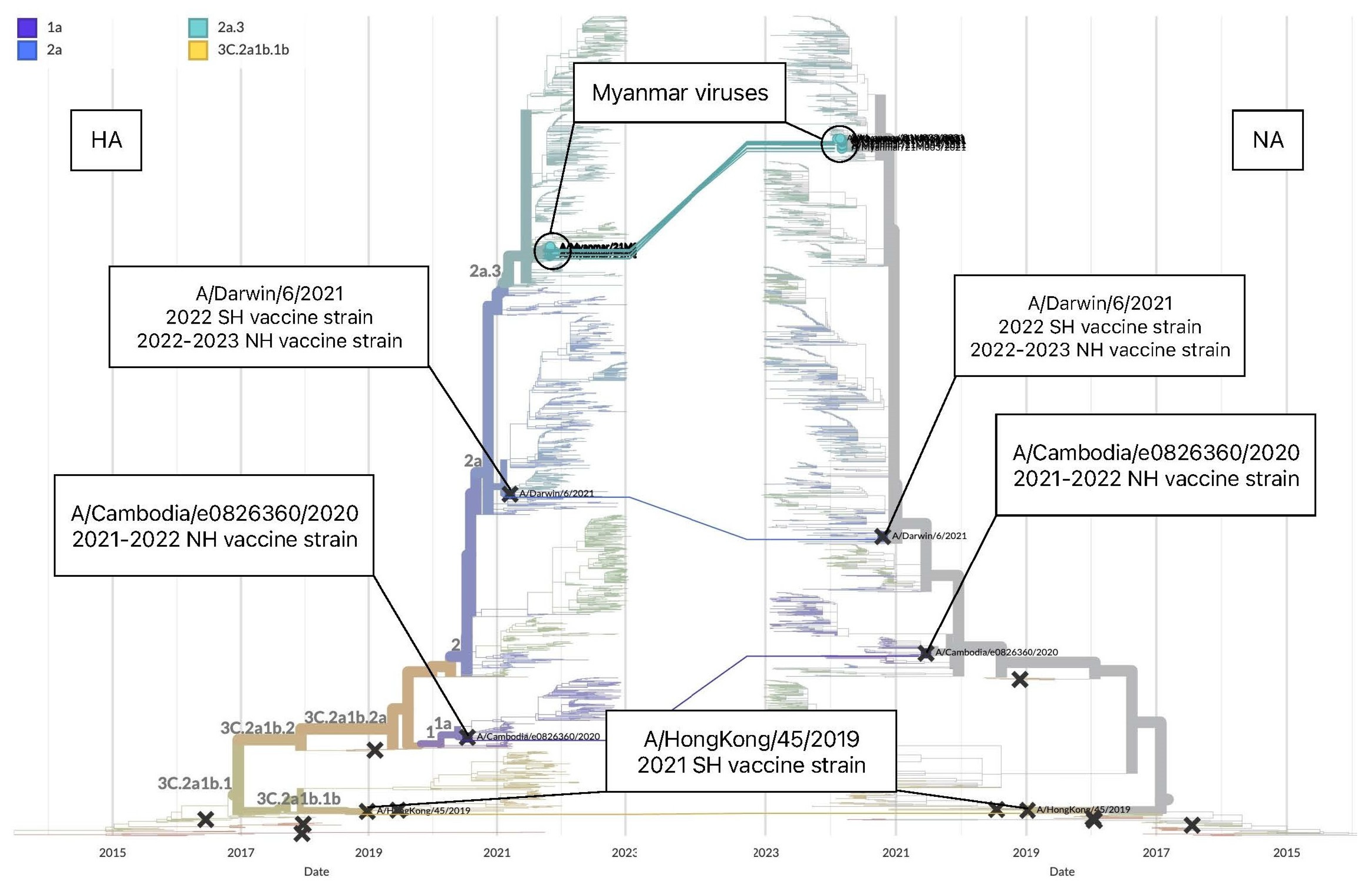
3.3.1. Genetic Analysis of Influenza A(H3N2)
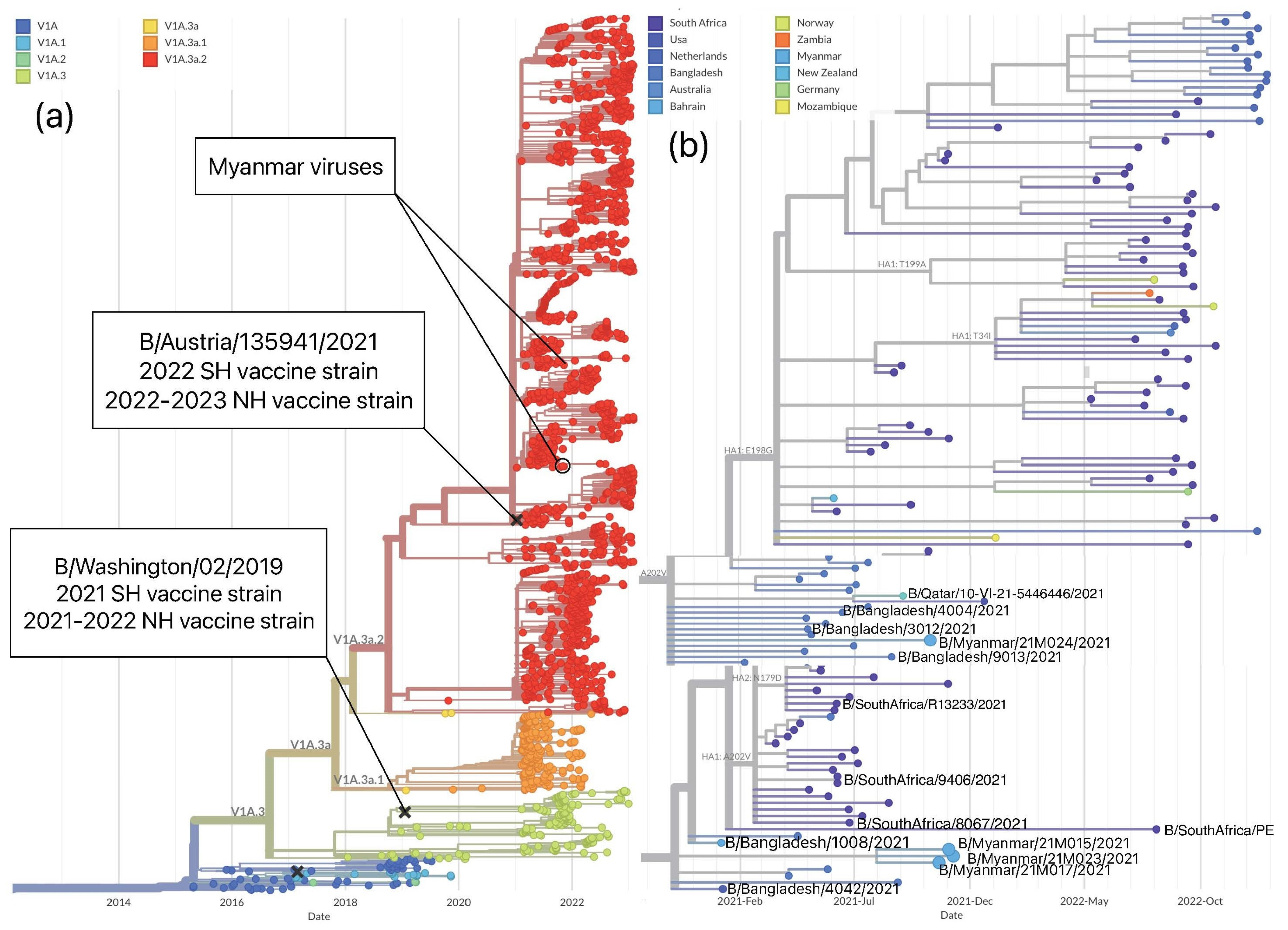
3.3.2. Genetic Analysis of Influenza B/Victoria
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization Influenza Seasonal. Available online: https://www.who.int/health-topics/influenza-seasonal#tab=tab_1 (accessed on 12 September 2022).
- WHO. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 July 2022).
- World Health Organization Influenza Laboratory Surveillance Information. Available online: https://app.powerbi.com/view?r=eyJrIjoiZTkyODcyOTEtZjA5YS00ZmI0LWFkZGUtODIxNGI5OTE3YjM0IiwidCI6ImY2MTBjMGI3LWJkMjQtNGIzOS04MTBiLTNkYzI4MGFmYjU5MCIsImMiOjh9 (accessed on 22 July 2022).
- Wagatsuma, K.; Koolhof, I.S.; Saito, R. Was the reduction in seasonal influenza transmission during 2020 attributable to non-pharmaceutical interventions to contain coronavirus disease 2019 in Japan? Viruses 2022, 14, 1417. [Google Scholar] [CrossRef] [PubMed]
- Win, S.M.K.; Saito, R.; Win, N.C.; Lasham, D.J.; Kyaw, Y.; Lin, N.; Thein, K.N.; Chon, I.; Odagiri, T.; Thein, W.; et al. Epidemic of influenza A(H1N1)pdm09 analyzed by full genome sequences and the first case of oseltamivir-resistant strain in Myanmar 2017. PLoS ONE 2020, 15, e0229601. [Google Scholar] [CrossRef] [Green Version]
- Dapat, C.; Saito, R.; Kyaw, Y.; Naito, M.; Hasegawa, G.; Suzuki, Y.; Dapat, I.C.; Zaraket, H.; Cho, T.M.; Li, D.; et al. Epidemiology of Human Influenza A and B Viruses in Myanmar from 2005 to 2007. Intervirology 2009, 52, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Htwe, K.T.Z.; Dapat, C.; Shobugawa, Y.; Odagiri, T.; Hibino, A.; Kondo, H.; Yagami, R.; Saito, T.; Takemae, N.; Tamura, T.; et al. Phylogeographic analysis of human influenza A and B viruses in Myanmar, 2010–2015. PLoS ONE 2019, 14, e0210550. [Google Scholar] [CrossRef] [PubMed]
- Dapat, C.; Suzuki, Y.; Saito, R.; Kyaw, Y.; Myint, Y.Y.; Lin, N.; Oo, H.N.; Oo, K.Y.; Win, N.; Naito, M.; et al. Rare Influenza A (H3N2) Variants with Reduced Sensitivity to Antiviral Drugs. Emerg. Infect. Dis. 2010, 16, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Dapat, C.; Saito, R.; Kyaw, Y.; Myint, Y.Y.; Oo, H.N.; Oo, K.Y.; Naito, M.; Hasegawa, G.; Dapat, I.C.; Suzuki, H. Delayed emergence of oseltamivir-resistant seasonal influenza A (H1N1) and pandemic influenza A(H1N1)pdm09 viruses in Myanmar. Influ. Other Respir. Viruses 2012, 7, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Phyu, W.W.; Saito, R.; Kyaw, Y.; Lin, N.; Win, S.M.K.; Win, N.C.; Di Ja, L.; Htwe, K.T.Z.; Aung, T.Z.; Tin, H.H.; et al. Evolutionary Dynamics of Whole-Genome Influenza A/H3N2 Viruses Isolated in Myanmar from 2015 to 2019. Viruses 2022, 14, 2414. [Google Scholar] [CrossRef]
- Phyu, W.W.; Saito, R.; Wagatsuma, K.; Abe, T.; Tin, H.H.; Pe, E.H.; Win, S.M.K.; Win, N.C.; Di Ja, L.; Tsuyoshi, S.; et al. Epidemiology and genetic analysis of SARS-CoV-2 in Myanmar during the community outbreaks in 2020. Viruses 2022, 14, 259. [Google Scholar] [CrossRef]
- Ministry of Health Myanmar COVID-19 Surveillance Dashboard. Available online: https://mohs.gov.mm/Main/content/new/list?pagenumber=1&pagesize=9 (accessed on 16 December 2022).
- Olsen, S.J.; Winn, A.K.; Budd, A.P.; Prill, M.M.; Steel, J.; Midgley, C.M.; Kniss, K.; Burns, E.; Rowe, T.; Foust, A.; et al. Changes in influenza and other respiratory virus activity during the COVID-19 pandemic—United States, 2020–2021. Morb. Mortal. Wkly. Rep. 2021, 70, 1013–1019. [Google Scholar] [CrossRef]
- World Health Organization Vaccines in Tropics and Subtropics. Available online: https://www.who.int/teams/global-influenza-programme/vaccines/vaccine-in-tropics-and-subtropics (accessed on 3 February 2023).
- Poritz, M.A.; Blaschke, A.J.; Byington, C.L.; Meyers, L.; Nilsson, K.; Jones, D.E.; Thatcher, S.A.; Robbins, T.; Lingenfelter, B.; Amiott, E.; et al. FilmArray, an automated nested multiplex PCR system for multi-pathogen detection: Development and application to respiratory tract infection. PLoS ONE 2011, 6, e26047. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Chen, H.; Apkarian, M.; Affrime, M.; Bock, B.; Kim, K.; Mukherjee, N.; Nolan, G.P.; McNeal, M.M. Performance of BioFire array or QuickVue influenza A + B test versus a validation qPCR assay for detection of influenza A during a volunteer A/California/2009/H1N1 challenge study. Virol. J. 2021, 18, 45. [Google Scholar] [CrossRef] [PubMed]
- Biofire Diagnostics, Biofire Respiratory Panel 2.1_De_Novo_Instructions_for_Use. Available online: https://www.biomerieux-diagnostics.com/biofirer-respiratory-21-plus-panel (accessed on 30 November 2022).
- Hoffmann, E.; Stech, J.; Guan, Y.; Webster, R.G.; Perez, D.R. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 2001, 146, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Saito, R.; Zaraket, H.; Dapat, C.; Caperig-Dapat, I.; Suzuki, H. Rapid and Specific Detection of Amantadine-Resistant Influenza A Viruses with a Ser31Asn Mutation by the Cycling Probe Method. J. Clin. Microbiol. 2010, 48, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Dapat, I.C.; Dapat, C.; Baranovich, T.; Suzuki, Y.; Kondo, H.; Shobugawa, Y.; Saito, R.; Suzuki, H.; The Japanese Influenza Collaborative Study Group. Genetic Characterization of Human Influenza Viruses in the Pandemic (2009–2010) and Post-Pandemic (2010–2011) Periods in Japan. PLoS ONE 2012, 7, e36455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, Y.; Saito, R.; Sato, I.; Zaraket, H.; Nishikawa, M.; Tamura, T.; Dapat, C.; Caperig-Dapat, I.; Baranovich, T.; Suzuki, T.; et al. Identification of Oseltamivir Resistance among Pandemic and Seasonal Influenza A (H1N1) Viruses by an His275Tyr Genotyping Assay Using the Cycling Probe Method. J. Clin. Microbiol. 2011, 49, 125–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Lin, X.; Wang, W.; Halpin, R.A.; Bera, J.; Stockwell, T.B.; Barr, I.G.; Wentworth, D.E. Universal Influenza B Virus Genomic Amplification Facilitates Sequencing, Diagnostics, and Reverse Genetics. J. Clin. Microbiol. 2014, 52, 1330–1337. [Google Scholar] [CrossRef] [Green Version]
- Mitchell, P.K.; Cronk, B.D.; Voorhees, I.E.H.; Rothenheber, D.; Anderson, R.R.; Chan, T.H.; Wasik, B.R.; Dubovi, E.J.; Parrish, C.R.; Goodman, L.B. Method comparison of targeted influenza A virus typing and whole-genome sequencing from respiratory specimens of companion animals. J. Veter- Diagn. Investig. 2020, 33, 191–201. [Google Scholar] [CrossRef]
- Wang, J.; Moore, N.; Edeng, Y.-M.; Eccles, D.A.; Hall, R.J. MinION nanopore sequencing of an influenza genome. Front. Microbiol. 2015, 6, 766. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Recommended Composition of Influenza Virus Vaccines for Use in the 2021 Southern Hemisphere Influenza Season; WHO: Geneva, Switzerland, 2020; pp. 1–9. Available online: https://www.who.int/publications/m/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2021_-southern-hemisphere-influenza-season (accessed on 25 September 2022).
- World Health Organization Recommended Composition of Influenza Virus Vaccines for Use in the 2022 Southern Hemisphere Influenza Season; WHO: Geneva, Switzerland, 2021; pp. 1–13. Available online: https://www.who.int/publications/m/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2022-southern-hemisphere-influenza-season (accessed on 24 September 2022).
- Lee, J.; Neher, R.; Bedford, T. Real-Time Tracking of Influenza A/H3N2 Evolution. Available online: https://nextstrain.org/flu/seasonal/h3n2/ha/2y (accessed on 17 February 2023).
- Lee, J.; Neher, R.; Bedford, T. Real-Time Tracking of Influenza B/Vic Evolution. Available online: https://nextstrain.org/flu/seasonal/vic/ha/2y (accessed on 17 February 2023).
- Worldwide Influenza Centre. The Francis Crick Institute Report Prepared for the WHO Annual Consultation on the Composition of Influenza Vaccine for the Southern Hemisphere 2022; Worldwide Influenza Centre: London, UK, 2021; pp. 1–95. Available online: https://www.crick.ac.uk/sites/default/files/2021-10/SH%20Sept%202021%20WIC%20web%20report%20v2.pdf (accessed on 25 November 2022).
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [Green Version]
- Worldwide Influenza Centre. The Francis Crick Institute Report Prepared for the WHO Annual Consultation on the Composition of Influenza Vaccines for the Southern Hemisphere 2023; Worldwide Influenza Centre: London, UK, 2022; pp. 1–132. Available online: https://www.crick.ac.uk/sites/default/files/2022-10/Crick%20report%20Sep2022%20for%20SH2023_to%20post.pdf (accessed on 25 November 2022).
- World Health Organization Recommended Composition of Influenza Virus Vaccines for Use in the 2021–2022 Northern Hemisphere Influenza Season; World Health Organization: Geneva, Switzerland, 2021; pp. 1–10. Available online: https://www.who.int/publications/i/item/recommended-composition-of-influenza-virus-vaccines-for-use-in-the-2021-2022-northern-hemisphere-influenza-season (accessed on 28 October 2022).
- World Health Organization Recommended Composition of Influenza Virus Vaccines for Use in the 2022–2023 Northern Hemisphere Influenza Season. Available online: https://www.who.int/news/item/25-02-2022-recommendations-announced-for-influenza-vaccine-composition-for-the-2022-2023-northern-hemisphere-influenza-season (accessed on 25 October 2022).
- Huang, W.; Li, X.; Tan, M.; Cheng, Y.; Chen, T.; Wei, H.; Zeng, X.; Wang, D. Epidemiological and virological surveillance of seasonal influenza viruses—China, 2020–2021. CDC Wkly. 2021, 3, 918–922. [Google Scholar] [CrossRef]
- Cowling, B.J.; Ali, S.T.; Ng, T.W.Y.; Tsang, T.K.; Li, J.C.M.; Fong, M.W.; Liao, Q.; Kwan, M.Y.; Lee, S.L.; Chiu, S.S.; et al. Impact assessment of non-pharmaceutical interventions against coronavirus disease 2019 and influenza in Hong Kong: An observational study. Lancet Public Health 2020, 5, e279–e288. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lee, H.; Song, K.-H.; Kim, E.S.; Park, J.S.; Jung, J.; Ahn, S.; Jeong, E.K.; Park, H.; Bin Kim, H. Impact of Public Health Interventions on Seasonal Influenza Activity During the COVID-19 Outbreak in Korea. Clin. Infect. Dis. 2020, 73, e132–e140. [Google Scholar] [CrossRef] [PubMed]
- Soo, R.J.J.; Chiew, C.J.; Ma, S.; Pung, R.; Lee, V. Decreased Influenza Incidence under COVID-19 Control Measures, Singapore. Emerg. Infect. Dis. 2020, 26, 1933–1935. [Google Scholar] [CrossRef] [PubMed]
- Lei, H.; Xu, M.; Wang, X.; Xie, Y.; Du, X.; Chen, T.; Yang, L.; Wang, D.; Shu, Y. Nonpharmaceutical Interventions Used to Control COVID-19 Reduced Seasonal Influenza Transmission in China. J. Infect. Dis. 2020, 222, 1780–1783. [Google Scholar] [CrossRef]
- Simon, B.; Pichon, M.; Valette, M.; Burfin, G.; Richard, M.; Lina, B.; Josset, L. Whole Genome Sequencing of A(H3N2) Influenza Viruses Reveals Variants Associated with Severity during the 2016–2017 Season. Viruses 2019, 11, 108. [Google Scholar] [CrossRef] [Green Version]
- Akhtar, Z.; Chowdhury, F.; Rahman, M.; Ghosh, P.K.; Ahmmed, K.; Islam, A.; Mott, J.A.; Davis, W. Seasonal influenza during the COVID-19 pandemic in Bangladesh. PLoS ONE 2021, 16, e0255646. [Google Scholar] [CrossRef]
- Melidou, A.; Gioula, G.; Exindari, M.; Ioannou, E.; Gkolfinopoulou, K.; Georgakopoulou, T.; Tsiodras, S.; Papa, A. Influenza A(H3N2) genetic variants in vaccinated patients in northern Greece. J. Clin. Virol. 2017, 94, 29–32. [Google Scholar] [CrossRef]
- Zost, S.J.; Parkhouse, K.; Gumina, M.E.; Kim, K.; Perez, S.D.; Wilson, P.C.; Treanor, J.J.; Sant, A.J.; Cobey, S.; Hensley, S.E. Contemporary H3N2 influenza viruses have a glycosylation site that alters binding of antibodies elicited by egg-adapted vaccine strains. Proc. Natl. Acad. Sci. USA 2017, 114, 12578–12583. [Google Scholar] [CrossRef] [Green Version]
- Rosu, M.E.; Lexmond, P.; Bestebroer, T.M.; Hauser, B.M.; Smith, D.J.; Herfst, S.; Fouchier, R.A.M. Substitutions near the HA receptor binding site explain the origin and major antigenic change of the B/Victoria and B/Yamagata lineages. Proc. Natl. Acad. Sci. USA 2022, 119, e2211616119. [Google Scholar] [CrossRef]
- Koel, B.F.; Burke, D.F.; Bestebroer, T.M.; van der Vliet, S.; Zondag, G.C.M.; Vervaet, G.; Skepner, E.; Lewis, N.S.; Spronken, M.I.J.; Russell, C.A.; et al. Substitutions near the Receptor Binding Site Determine Major Antigenic Change during Influenza Virus Evolution. Science 2013, 342, 976–979. [Google Scholar] [CrossRef]
- Chen, R.; Holmes, E.C. The Evolutionary Dynamics of Human Influenza B Virus. J. Mol. Evol. 2008, 66, 655–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, S.; Chadha, M.; Al Mamun, A.; Rahman, M.; Sturm-Ramirez, K.; Chittaganpitch, M.; Pattamadilok, S.; Olsen, S.J.; Sampurno, O.D.; Setiawaty, V.; et al. Influenza seasonality and vaccination timing in tropical and subtropical areas of southern and south-eastern Asia. Bull. World Health Organ. 2014, 92, 318–330. [Google Scholar] [CrossRef] [PubMed]
- Ampofo, W.K.; Azziz-Baumgartner, E.; Bashir, U.; Cox, N.J.; Fasce, R.; Giovanni, M.; Grohmann, G.; Huang, S.; Katz, J.; Mironenko, A.; et al. Strengthening the influenza vaccine virus selection and development process: Report of the 3rd WHO Informal Consultation for Improving Influenza Vaccine Virus Selection held at WHO headquarters, Geneva, Switzerland, 1–3 April 2014. Vaccine 2015, 33, 4368–4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, H.; Shobugawa, Y.; Hibino, A.; Yagami, R.; Dapat, C.; Okazaki, M.; Otsuka, T.; Fujii, K.; Hassan, M.R.; Saito, R. Influenza Virus Shedding in Laninamivir-Treated Children upon Returning to School. Tohoku J. Exp. Med. 2016, 238, 113–121. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type (Subtype/Lineage) | A(H3N2) (n = 13) | B/Victoria (n = 3) |
|---|---|---|
| Variables | ||
| Age, median [min–max] (years) | 2 [0.5–7] | 2 [1–4] |
| Gender, male (%) | 7 (53.8) | 3 (100) |
| Symptoms, presence (%) | ||
| Cough | 13 (100) | 3 (100) |
| Fever ≥ 37.5 °C | 5 (38.5) | 2 (66.6) |
| Rhinorrhea | 10 (76.9) | 2 (66.6) |
| Headache | 0 (0.0) | 1 (33.3) |
| Diarrhea | 0 (0.0) | 1 (33.3) |
| Influenza vaccination | ||
| Presence (%) | 1 (7.7) | 0 (0.0) |
| Amino Acid Substitutions | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Segment | HA | |||||||||||
| Vaccine strain | Clade | 53 | 83 | 94 | 96 | 128 | 131 | 135 | 137 | 138 | 156 | 159 * |
| A/Hong Kong/45/2019 | 3C.2a1b.1b | D | K | Y | N | A | T | K | F | S | H | Y |
| Myanmar viruses 2021 | 2a.3 | N | E | N | S | T | K | T | S | A | S | N |
| Segment | HA | |||||||||||
| Vaccine strain | Clade | 160 * | 164 | 186 | 190 | 192 | 195 | 312 | 378 | 522 | 529 | |
| A/Hong Kong/45/2019 | 3C.2a1b.1b | T | L | G | D | I | Y | N | N | I | V | |
| Myanmar viruses 2021 | 2a.3 | I | Q | D | N | F | F | S | S | M | I | |
| Amino Acid Substitutions | ||||||
|---|---|---|---|---|---|---|
| Segment | HA | |||||
| Vaccine strain | Clade | 53 | 96 | 192 | 225 | 378 |
| A/Darwin/6/2021 | 2a | D | N | I | G | N |
| Myanmar viruses 2021 | 2a.3 | N | S | F | D | S |
| Amino Acid Substitutions | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Segment | HA | ||||||||
| Vaccine strain | Clade | 127 | 133 | 144 | 150 | 181 | 194 | 203 | 276 |
| B/Washington/02/2019 | V1A.3 | A | R | P | N | G | N | K | R |
| Myanmar viruses 2021 | V1A.3a.2 | T | G | L | K | E | E/D | R | K |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chon, I.; Saito, R.; Kyaw, Y.; Aye, M.M.; Setk, S.; Phyu, W.W.; Wagatsuma, K.; Li, J.; Sun, Y.; Otoguro, T.; et al. Whole-Genome Analysis of Influenza A(H3N2) and B/Victoria Viruses Detected in Myanmar during the COVID-19 Pandemic in 2021. Viruses 2023, 15, 583. https://doi.org/10.3390/v15020583
Chon I, Saito R, Kyaw Y, Aye MM, Setk S, Phyu WW, Wagatsuma K, Li J, Sun Y, Otoguro T, et al. Whole-Genome Analysis of Influenza A(H3N2) and B/Victoria Viruses Detected in Myanmar during the COVID-19 Pandemic in 2021. Viruses. 2023; 15(2):583. https://doi.org/10.3390/v15020583
Chicago/Turabian StyleChon, Irina, Reiko Saito, Yadanar Kyaw, Moe Myat Aye, Swe Setk, Wint Wint Phyu, Keita Wagatsuma, Jiaming Li, Yuyang Sun, Teruhime Otoguro, and et al. 2023. "Whole-Genome Analysis of Influenza A(H3N2) and B/Victoria Viruses Detected in Myanmar during the COVID-19 Pandemic in 2021" Viruses 15, no. 2: 583. https://doi.org/10.3390/v15020583