
Diverse Marine T4-like Cyanophage Communities Are Primarily Comprised of Low-Abundance Species Including Species with Distinct Seasonal, Persistent, Occasional, or Sporadic Dynamics

Abstract
:1. Introduction
2. Materials and Methods
2.1. ANI-Informed Classification of Cyanophage Species
2.2. Sample Collection and Processing
2.3. Assembly and Identification of gp43 Sequences
2.4. Clustering and Mapping
2.5. Cyanophage Community Analysis
2.6. Seasonal Dynamics
2.7. Network Analysis
2.8. Phylogenetic Analysis
2.9. Variance Partitioning
3. Results
3.1. Classification of T4-like Cyanophage Species
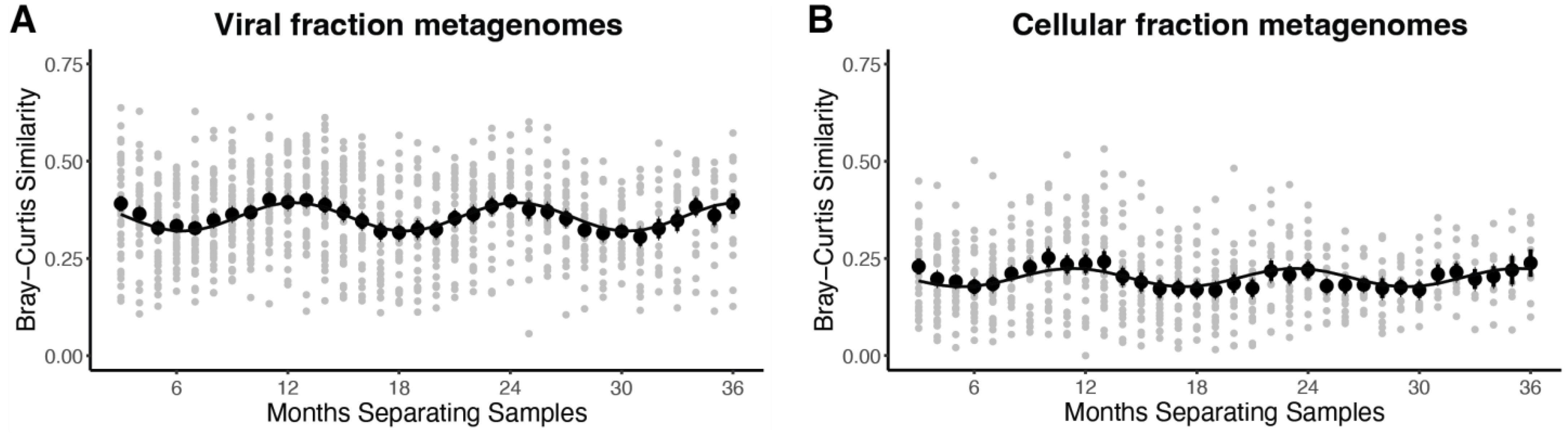
3.2. T4-like Cyanophage Community Composition and Whole Community Dynamics
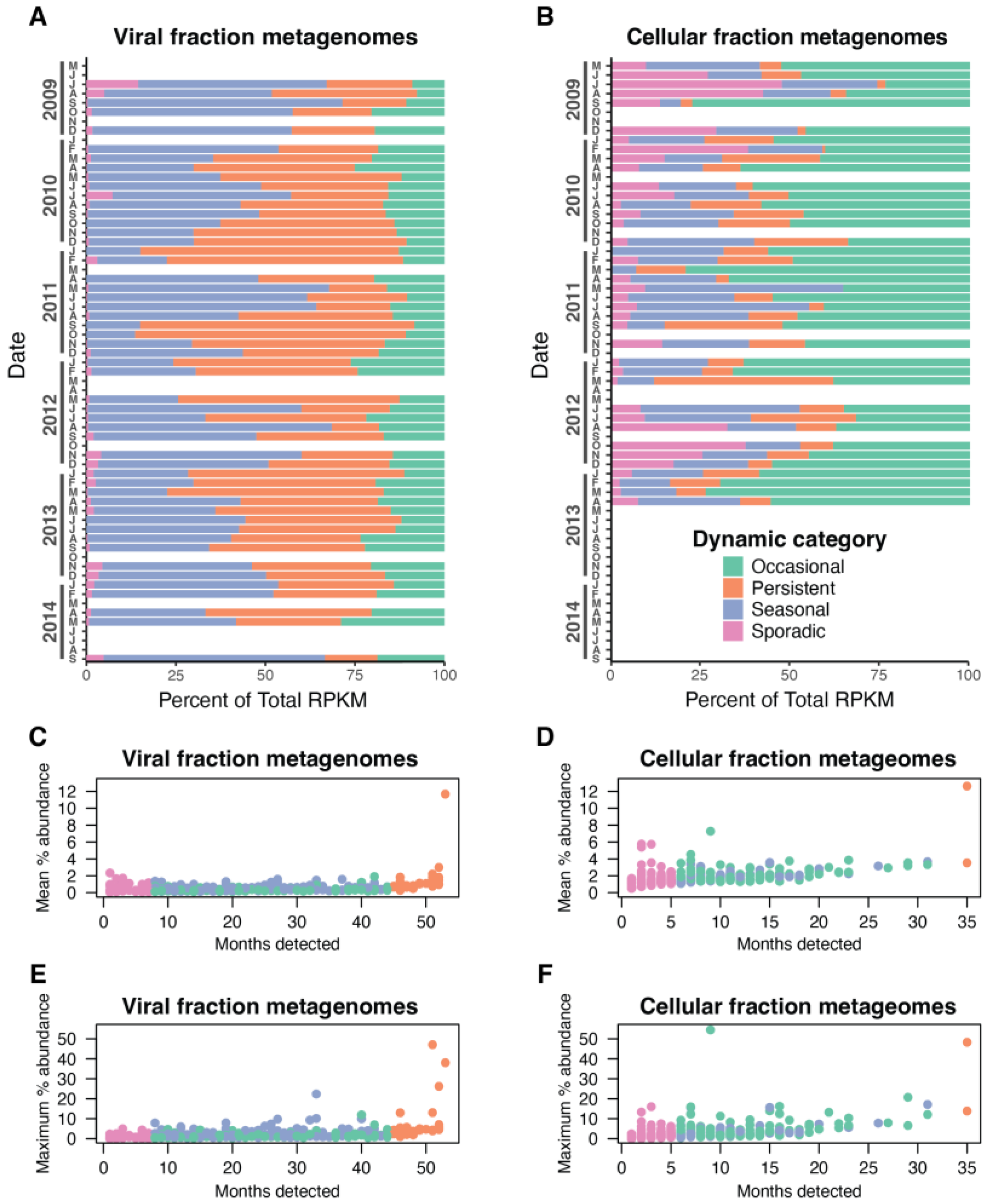
3.3. Dynamic Patterns of Individual Cyanophage Species
3.4. Community Network Analysis
3.5. Phylogenetic Relatedness of Cyanophage Dynamic Phenotypes
3.6. Variance Partitioning Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sullivan, M.B.; Waterbury, J.B.; Chisholm, S.W. Cyanophage Infecting the Oceanic Cyanobacterium. Nature 2003, 424, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Andrews-Pfannkoch, C.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The Sorcerer II Global Ocean Sampling Expedition: Metagenomic Characterization of Viruses within Aquatic Microbial Samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef]
- Weitz, J.S.; Wilhelm, S.W. Ocean Viruses and Their Effects on Microbial Communities and Biogeochemical Cycles. F1000 Biol. Rep. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, R.J.; Breitbart, M.; Lomas, M.W.; Carlson, C.A. Ocean Time-Series Reveals Recurring Seasonal Patterns of Virioplankton Dynamics in the Northwestern Sargasso Sea. ISME J. 2012, 6, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Hevroni, G.; Flores-Uribe, J.; Béjà, O.; Philosof, A. Seasonal and Diel Patterns of Abundance and Activity of Viruses in the Red Sea. Proc. Natl. Acad. Sci. USA 2020, 117, 29738–29747. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Bouvier, T.; Weinbauer, M.G.; Thingstad, T.F. Trade-Offs between Competition and Defense Specialists among Unicellular Planktonic Organisms: The “Killing the Winner” Hypothesis Revisited. Microbiol. Mol. Biol. Rev. 2010, 74, 42–57. [Google Scholar] [CrossRef] [Green Version]
- Thingstad, T.F. Elements of a Theory for the Mechanisms Controlling Abundance, Diversity, and Biogeochemical Role of Lytic Bacterial Viruses in Aquatic Systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Rodriguez-Brito, B.; Li, L.; Wegley, L.; Furlan, M.; Angly, F.; Breitbart, M.; Buchanan, J.; Desnues, C.; Dinsdale, E.; Edwards, R.; et al. Viral and Microbial Community Dynamics in Four Aquatic Environments. ISME J. 2010, 4, 739–751. [Google Scholar] [CrossRef]
- Marston, M.F.; Pierciey, F.J.; Shepard, A.; Gearin, G.; Qi, J.; Yandava, C.; Schuster, S.C.; Henn, M.R.; Martiny, J.B.H. Rapid Diversification of Coevolving Marine Synechococcus and a Virus. Proc. Natl. Acad. Sci. USA 2012, 109, 4544–4549. [Google Scholar] [CrossRef] [Green Version]
- Ahlgren, N.A.; Perelman, J.N.; Yeh, Y.; Fuhrman, J.A. Multi-year Dynamics of Fine-scale Marine Cyanobacterial Populations Are More Strongly Explained by Phage Interactions than Abiotic, Bottom-up Factors. Environ. Microbiol. 2019, 21, 2948–2963. [Google Scholar] [CrossRef]
- Ignacio-Espinoza, J.C.; Ahlgren, N.A.; Fuhrman, J.A. Long-Term Stability and Red Queen-like Strain Dynamics in Marine Viruses. Nat. Microbiol. 2020, 5, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Rohwer, F. Here a Virus, There a Virus, Everywhere the Same Virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef]
- Finke, J.F.; Suttle, C.A. The Environment and Cyanophage Diversity: Insights From Environmental Sequencing of DNA Polymerase. Front. Microbiol. 2019, 10, 167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brum, J.R.; Hurwitz, B.L.; Schofield, O.; Ducklow, H.W.; Sullivan, M.B. Seasonal Time Bombs: Dominant Temperate Viruses Affect Southern Ocean Microbial Dynamics. ISME J. 2016, 10, 437–449. [Google Scholar] [CrossRef] [PubMed]
- Güemes, A.G.C.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners in the Game of Life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef]
- Needham, D.M.; Sachdeva, R.; Fuhrman, J.A. Ecological Dynamics and Co-Occurrence among Marine Phytoplankton, Bacteria and Myoviruses Shows Microdiversity Matters. ISME J. 2017, 11, 1614–1629. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A.; Cram, J.A.; Needham, D.M. Marine Microbial Community Dynamics and Their Ecological Interpretation. Nat. Rev. Microbiol. 2015, 13, 133–146. [Google Scholar] [CrossRef]
- Marston, M.F.; Amrich, C.G. Recombination and Microdiversity in Coastal Marine Cyanophages. Environ. Microbiol. 2009, 11, 2893–2903. [Google Scholar] [CrossRef]
- Clasen, J.; Hanson, C.; Ibrahim, Y.; Weihe, C.; Marston, M.; Martiny, J. Diversity and Temporal Dynamics of Southern California Coastal Marine Cyanophage Isolates. Aquat. Microb. Ecol. 2013, 69, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Needham, D.M.; Chow, C.-E.T.; Cram, J.A.; Sachdeva, R.; Parada, A.; Fuhrman, J.A. Short-Term Observations of Marine Bacterial and Viral Communities: Patterns, Connections and Resilience. ISME J. 2013, 7, 1274–1285. [Google Scholar] [CrossRef] [Green Version]
- Pagarete, A.; Chow, C.-E.T.; Johannessen, T.; Fuhrman, J.A.; Thingstad, T.F.; Sandaa, R.A. Strong Seasonality and Interannual Recurrence in Marine Myovirus Communities. Appl. Environ. Microbiol. 2013, 79, 6253–6259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, M.F.; Martiny, J.B.H. Genomic Diversification of Marine Cyanophages into Stable Ecotypes: Cyanophage Diversification into Ecotypes. Environ. Microbiol. 2016, 18, 4240–4253. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-E.T.; Fuhrman, J.A. Seasonality and Monthly Dynamics of Marine Myovirus Communities: Marine Myovirus Community Dynamics at SPOT. Environ. Microbiol. 2012, 14, 2171–2183. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Solonenko, S.A.; Ignacio-Espinoza, J.C.; LaButti, K.; Copeland, A.; Sudek, S.; Maitland, A.; Chittick, L.; dos Santos, F.; Weitz, J.S.; et al. Genomic Differentiation among Wild Cyanophages despite Widespread Horizontal Gene Transfer. BMC Genom. 2016, 17, 930. [Google Scholar] [CrossRef] [Green Version]
- Zborowsky, S.; Lindell, D. Resistance in Marine Cyanobacteria Differs against Specialist and Generalist Cyanophages. Proc. Natl. Acad. Sci. USA 2019, 116, 16899–16908. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.-E.T.; Kim, D.Y.; Sachdeva, R.; Caron, D.A.; Fuhrman, J.A. Top-down Controls on Bacterial Community Structure: Microbial Network Analysis of Bacteria, T4-like Viruses and Protists. ISME J. 2014, 8, 816–829. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Ignacio-Espinoza, J.C.; Gregory, A.C.; Poulos, B.T.; Weitz, J.S.; Hugenholtz, P.; Sullivan, M.B. Viral Tagging Reveals Discrete Populations in Synechococcus Viral Genome Sequence Space. Nature 2014, 513, 242–245. [Google Scholar] [CrossRef]
- Marston, M.F.; Taylor, S.; Sme, N.; Parsons, R.J.; Noyes, T.J.E.; Martiny, J.B.H. Marine Cyanophages Exhibit Local and Regional Biogeography: Biogeography of Marine Cyanophages. Environ. Microbiol. 2013, 15, 1452–1463. [Google Scholar] [CrossRef]
- Cram, J.A.; Chow, C.-E.T.; Sachdeva, R.; Needham, D.M.; Parada, A.E.; Steele, J.A.; Fuhrman, J.A. Seasonal and Interannual Variability of the Marine Bacterioplankton Community throughout the Water Column over Ten Years. ISME J. 2015, 9, 563–580. [Google Scholar] [CrossRef] [Green Version]
- Yeh, Y.; McNichol, J.; Needham, D.M.; Fichot, E.B.; Berdjeb, L.; Fuhrman, J.A. Comprehensive single-PCR 16S and 18S rRNA Community Analysis Validated with Mock Communities, and Estimation of Sequencing Bias against 18S. Environ. Microbiol. 2021, 23, 3240–3250. [Google Scholar] [CrossRef]
- Rocap, G.; Distel, D.L.; Waterbury, J.B.; Chisholm, S.W. Resolution of Prochlorococcus and Synechococcus Ecotypes by Using 16S-23S Ribosomal DNA Internal Transcribed Spacer Sequences. Appl. Environ. Microbiol. 2002, 68, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Lavin, P.; Gomez, P.; Gonzalez, B.; Ulloa, O. Diversity of the Marine Picocyanobacteria Prochlorococcus and Synechococcus Assessed by Terminal Restriction Fragment Length Polymorphisms of 16S-23S RRNA Internal Transcribed Spacer Sequences. Rev. Chil. Hist. Nat. 2008, 81, 515–531. [Google Scholar] [CrossRef]
- Eren, A.M.; Morrison, H.G.; Lescault, P.J.; Reveillaud, J.; Vineis, J.H.; Sogin, M.L. Minimum Entropy Decomposition: Unsupervised Oligotyping for Sensitive Partitioning of High-Throughput Marker Gene Sequences. ISME J. 2015, 9, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Chisholm, S.W. Code and Context: Prochlorococcus as a Model for Cross-Scale Biology. Trends Microbiol. 2007, 15, 398–407. [Google Scholar] [CrossRef]
- Biller, S.J.; Berube, P.M.; Lindell, D.; Chisholm, S.W. Prochlorococcus: The Structure and Function of Collective Diversity. Nat. Rev. Microbiol. 2015, 13, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Sohm, J.A.; Ahlgren, N.A.; Thomson, Z.J.; Williams, C.; Moffett, J.W.; Saito, M.A.; Webb, E.A.; Rocap, G. Co-Occurring Synechococcus Ecotypes Occupy Four Major Oceanic Regimes Defined by Temperature, Macronutrients and Iron. ISME J. 2016, 10, 333–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bushnell. Brian BBMap. Available online: sourceforge.net/projects/bbmap/ (accessed on 10 February 2022).
- Li, H. BFC: Correcting Illumina Sequencing Errors. Bioinformatics 2015, 31, 2885–2887. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining Viral Signal from Microbial Genomic Data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Ren, J.; Ahlgren, N.A.; Lu, Y.Y.; Fuhrman, J.A.; Sun, F. VirFinder: A Novel k-Mer Based Tool for Identifying Viral Sequences from Assembled Metagenomic Data. Microbiome 2017, 5, 69. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Bolton, E.E.; Brister, J.R.; Canese, K.; Chan, J.; Comeau, D.C.; Connor, R.; Funk, K.; Kelly, C.; Kim, S.; et al. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2022, 50, D20–D26. [Google Scholar] [CrossRef]
- Wang, Q.; Fish, J.A.; Gilman, M.; Sun, Y.; Brown, C.T.; Tiedje, J.M.; Cole, J.R. Xander: Employing a Novel Method for Efficient Gene-Targeted Metagenomic Assembly. Microbiome 2015, 3, 32. [Google Scholar] [CrossRef] [Green Version]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A Versatile Open Source Tool for Metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Simpson, G.L.; Kindt, R.; Legendre, P.; Minchin, P.R. Vegan: Community Ecology Package. R Package Version 2. 2020. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 20 January 2022).
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.R-project.org/ (accessed on 10 March 2022).
- Wood, S.N. Stable and Efficient Multiple Smoothing Parameter Estimation for Generalized Additive Models. J. Am. Stat. Assoc. 2004, 99, 673–686. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.N. Generalized Additive Models An Introduction with R, 1st ed.; Chapman and Hall/CRC: New York, NY, USA, 2006; ISBN 978-0-429-09315-9. [Google Scholar]
- Wood, S.N.; Scheipl, F. Gamm4. Available online: https://cran.r-project.org/web/packages/gamm4/index.html (accessed on 10 March 2022).
- Ferguson, C.A.; Carvalho, L.; Scott, E.M.; Bowman, A.W.; Kirika, A. Assessing Ecological Responses to Environmental Change Using Statistical Models: Methods for Assessing Trends and Seasonality. J. Appl. Ecol. 2007, 45, 193–203. [Google Scholar] [CrossRef]
- Storey, J.D.; Bass, A.J.; Dabney, A.; Robinson, D. qvalue: Q-Value Estimation for False Discovery Rate Control. R Package Version 2.30.0. 2022. Available online: http://github.com/jdstorey/qvalue (accessed on 23 January 2023).
- Xia, L.C.; Steele, J.A.; Cram, J.A.; Cardon, Z.G.; Simmons, S.L.; Vallino, J.J.; Fuhrman, J.A.; Sun, F. Extended Local Similarity Analysis (ELSA) of Microbial Community and Other Time Series Data with Replicates. BMC Syst. Biol. 2011, 5, S15–2504. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Bininda-Emonds, O.R. TransAlign: Using Amino Acids to Facilitate the Multiple Alignment of Protein-Coding DNA Sequences. BMC Bioinform. 2005, 6, 156. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Schwartz, T.; Pickett, B.E.; He, S.; Klem, E.B.; Scheuermann, R.H.; Passarotti, M.; Kaufman, S.; O’Leary, M.A. A RESTful API for Access to Phylogenetic Tools via the CIPRES Science Gateway. Evol. Bioinform. 2015, 11, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Peres-Neto, P.R.; Legendre, P.; Dray, S.; Borcard, D. Variation Partitioning of Species Data Matrices: Estimation and Comparison of Fractions. Ecology 2006, 87, 2614–2625. [Google Scholar] [CrossRef] [PubMed]
- Larkin, A.A.; Moreno, A.R.; Fagan, A.J.; Fowlds, A.; Ruiz, A.; Martiny, A.C. Persistent El Niño Driven Shifts in Marine Cyanobacteria Populations. PLoS ONE 2020, 15, e0238405. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.-E.T.; Sachdeva, R.; Cram, J.A.; Steele, J.A.; Needham, D.M.; Patel, A.; Parada, A.E.; Fuhrman, J.A. Temporal Variability and Coherence of Euphotic Zone Bacterial Communities over a Decade in the Southern California Bight. ISME J. 2013, 7, 2259–2273. [Google Scholar] [CrossRef] [Green Version]
- Flores, C.O.; Meyer, J.R.; Valverde, S.; Farr, L.; Weitz, J.S. Statistical Structure of Host–Phage Interactions. Proc. Natl. Acad. Sci. USA 2011, 108. [Google Scholar] [CrossRef] [Green Version]
- Flores, C.O.; Valverde, S.; Weitz, J.S. Multi-Scale Structure and Geographic Drivers of Cross-Infection within Marine Bacteria and Phages. ISME J. 2013, 7, 520–532. [Google Scholar] [CrossRef] [Green Version]
- Avrani, S.; Wurtzel, O.; Sharon, I.; Sorek, R.; Lindell, D. Genomic Island Variability Facilitates Prochlorococcus–Virus Coexistence. Nature 2011, 474, 604–608. [Google Scholar] [CrossRef]
- Dekel-Bird, N.P.; Avrani, S.; Sabehi, G.; Pekarsky, I.; Marston, M.F.; Kirzner, S.; Lindell, D. Diversity and Evolutionary Relationships of T 7-like Podoviruses Infecting Marine Cyanobacteria. Environ. Microbiol. 2013, 15, 1476–1491. [Google Scholar] [CrossRef]
- Garza, D.; Suttle, C.A. The Effect of Cyanophages on the Mortality of Synechococcus Spp. and Selection for UV Resistant Viral Communities. Microb. Ecol. 1998, 36, 281–292. [Google Scholar] [CrossRef]
- Suttle, C.A.; Chen, F. Mechanisms and Rates of Decay of Marine Viruses in Seawater. Appl. Environ. Microbiol. 1992, 58, 3721–3729. [Google Scholar] [CrossRef] [Green Version]
- Chow, C.-E.T.; Suttle, C.A. Biogeography of Viruses in the Sea. Annu. Rev. Virol. 2015, 2, 41–66. [Google Scholar] [CrossRef]
- Cheng, K.; Zhao, Y.; Du, X.; Zhang, Y.; Lan, S.; Zhengli, S. Solar Radiation-Driven Decay of Cyanophage Infectivity, and Photoreactivation of the Cyanophage by Host Cyanobacteria. Aquat. Microb. Ecol. 2007, 48, 13–18. [Google Scholar] [CrossRef] [Green Version]
- Maidanik, I.; Kirzner, S.; Pekarski, I.; Arsenieff, L.; Tahan, R.; Carlson, M.C.G.; Shitrit, D.; Baran, N.; Goldin, S.; Weitz, J.S.; et al. Cyanophages from a Less Virulent Clade Dominate over Their Sister Clade in Global Oceans. ISME J. 2022, 16, 2169–2180. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.R.; Post, A.F.; Rocap, G.; Chisholm, S.W. Utilization of Different Nitrogen Sources by the Marine Cyanobacteria Prochlorococcus and Synechococcus. Limnol. Oceanogr. 2002, 47, 989–996. [Google Scholar] [CrossRef]
- Johnson, Z.I.; Zinser, E.R.; Coe, A.; McNulty, N.P.; Woodward, E.M.S.; Chisholm, S.W. Niche Partitioning Among Prochlorococcus Ecotypes Along Ocean-Scale Environmental Gradients. Science 2006, 311, 1737–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiny, A.C.; Tai, A.P.K.; Veneziano, D.; Primeau, F.; Chisholm, S.W. Taxonomic Resolution, Ecotypes and the Biogeography of Prochlorococcus. Environ. Microbiol. 2009, 11, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Worden, A.; Binder, B. Application of Dilution Experiments for Measuring Growth and Mortality Rates among Prochlorococcus and Synechococcus Populations in Oligotrophic Environments. Aquat. Microb. Ecol. 2003, 30, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Apple, J.K.; Strom, S.L.; Palenik, B.; Brahamsha, B. Variability in Protist Grazing and Growth on Different Marine Synechococcus Isolates. Appl. Environ. Microbiol. 2011, 77, 3074–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilo-Ferretjans, M.D.M.; Bosch, R.; Puxty, R.J.; Latva, M.; Zadjelovic, V.; Chhun, A.; Sousoni, D.; Polin, M.; Scanlan, D.J.; Christie-Oleza, J.A. Pili Allow Dominant Marine Cyanobacteria to Avoid Sinking and Evade Predation. Nat. Commun. 2021, 12, 1857. [Google Scholar] [CrossRef]
- Paz-Yepes, J.; Brahamsha, B.; Palenik, B. Role of a Microcin-C–like Biosynthetic Gene Cluster in Allelopathic Interactions in Marine Synechococcus. Proc. Natl. Acad. Sci. USA 2013, 110, 12030–12035. [Google Scholar] [CrossRef] [Green Version]
- Baran, N.; Carlson, M.C.G.; Sabehi, G.; Peleg, M.; Kondratyeva, K.; Pekarski, I.; Lindell, D. Widespread yet Persistent Low Abundance of TIM5 -like Cyanophages in the Oceans. Environ. Microbiol. 2022, 24, 6476–6492. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Number of Connections |
|---|---|
| Host Ecotype—Viruses in Cellular Metagenomes | 38 |
| Host Ecotype—Viruses in Viral Fraction Metagenomes | 11 |
| Host ASV—Viruses in Cellular Metagenomes | 223 |
| Host ASV—Viruses in Viral Fraction Metagenomes | 58 |
| Host ASV—Host ASV | 1524 |
| Viruses in Viral Fraction Metagenomes—Viruses in Viral Fraction Metagenomes | 905 |
| Viruses in Viral Fraction Metagenomes—Viruses in Cellular Metagenomes | 844 |
| Viruses in Cellular Metagenomes—Viruses in Cellular Metagenomes | 936 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dart, E.; Fuhrman, J.A.; Ahlgren, N.A. Diverse Marine T4-like Cyanophage Communities Are Primarily Comprised of Low-Abundance Species Including Species with Distinct Seasonal, Persistent, Occasional, or Sporadic Dynamics. Viruses 2023, 15, 581. https://doi.org/10.3390/v15020581
Dart E, Fuhrman JA, Ahlgren NA. Diverse Marine T4-like Cyanophage Communities Are Primarily Comprised of Low-Abundance Species Including Species with Distinct Seasonal, Persistent, Occasional, or Sporadic Dynamics. Viruses. 2023; 15(2):581. https://doi.org/10.3390/v15020581
Chicago/Turabian StyleDart, Emily, Jed A. Fuhrman, and Nathan A. Ahlgren. 2023. "Diverse Marine T4-like Cyanophage Communities Are Primarily Comprised of Low-Abundance Species Including Species with Distinct Seasonal, Persistent, Occasional, or Sporadic Dynamics" Viruses 15, no. 2: 581. https://doi.org/10.3390/v15020581