
Fragment-Based Approaches Identified Tecovirimat-Competitive Novel Drug Candidate for Targeting the F13 Protein of the Monkeypox Virus

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Methods
2.1. Modeling and Preparation of F13 Protein
2.2. Fragment Library Preparation
2.3. Virtual Screening of Fragment Library
2.4. Molecular Docking via AutoDock Vina
2.5. Analysis of Computational Pharmacokinetics
2.6. Explicit Solvent Molecular Dynamics Simulation
2.7. Essential Dynamics Analysis via Principal Components
3. Results
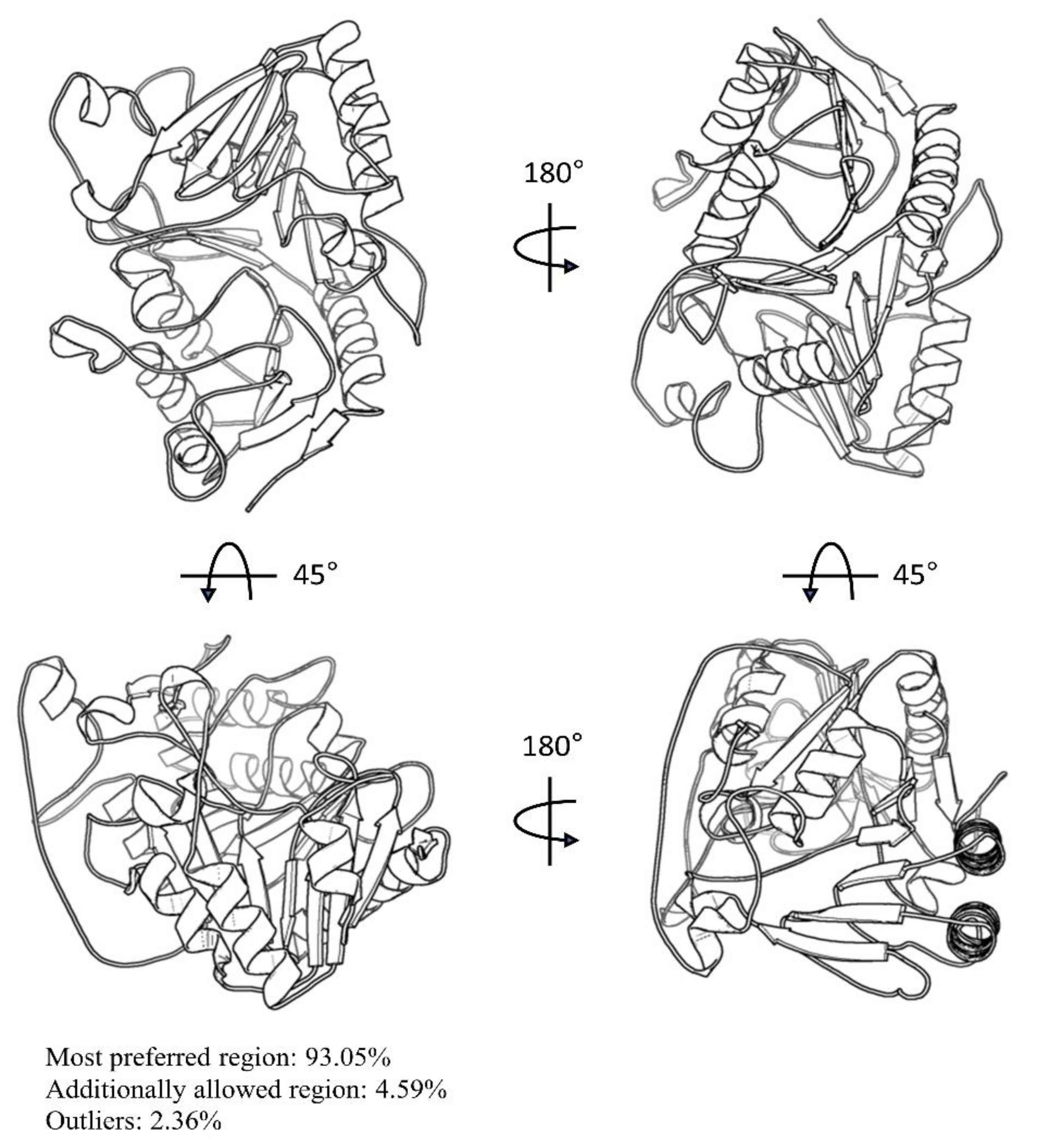
3.1. Preparation of F13 Protein
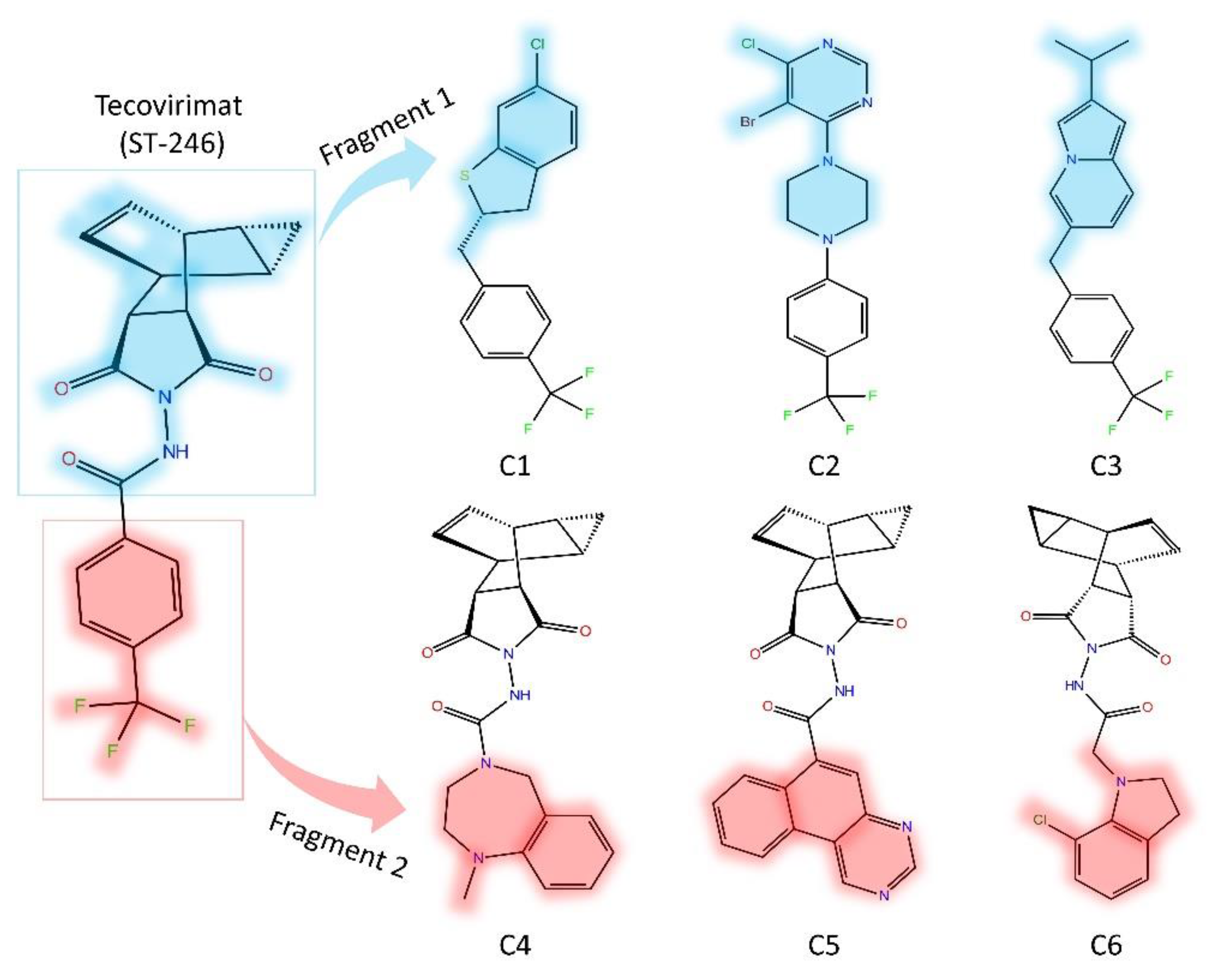
3.2. Fragment-Based Compounds Library Development Using ST-246 Molecular Properties
3.3. Virtual Screening and Selection of Top Hits
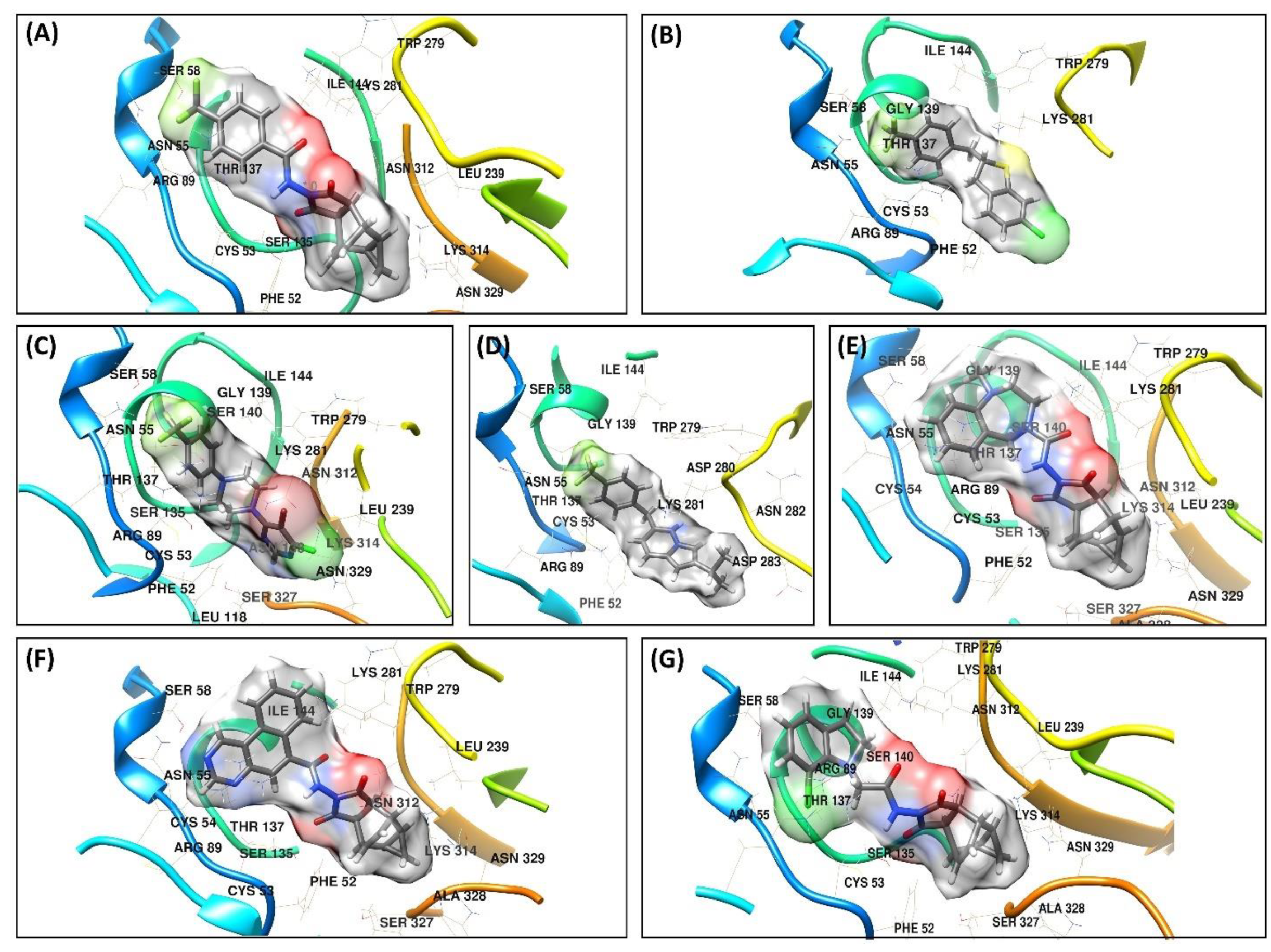
3.4. Molecular Docking Analysis
3.5. Computational Pharmacokinetics
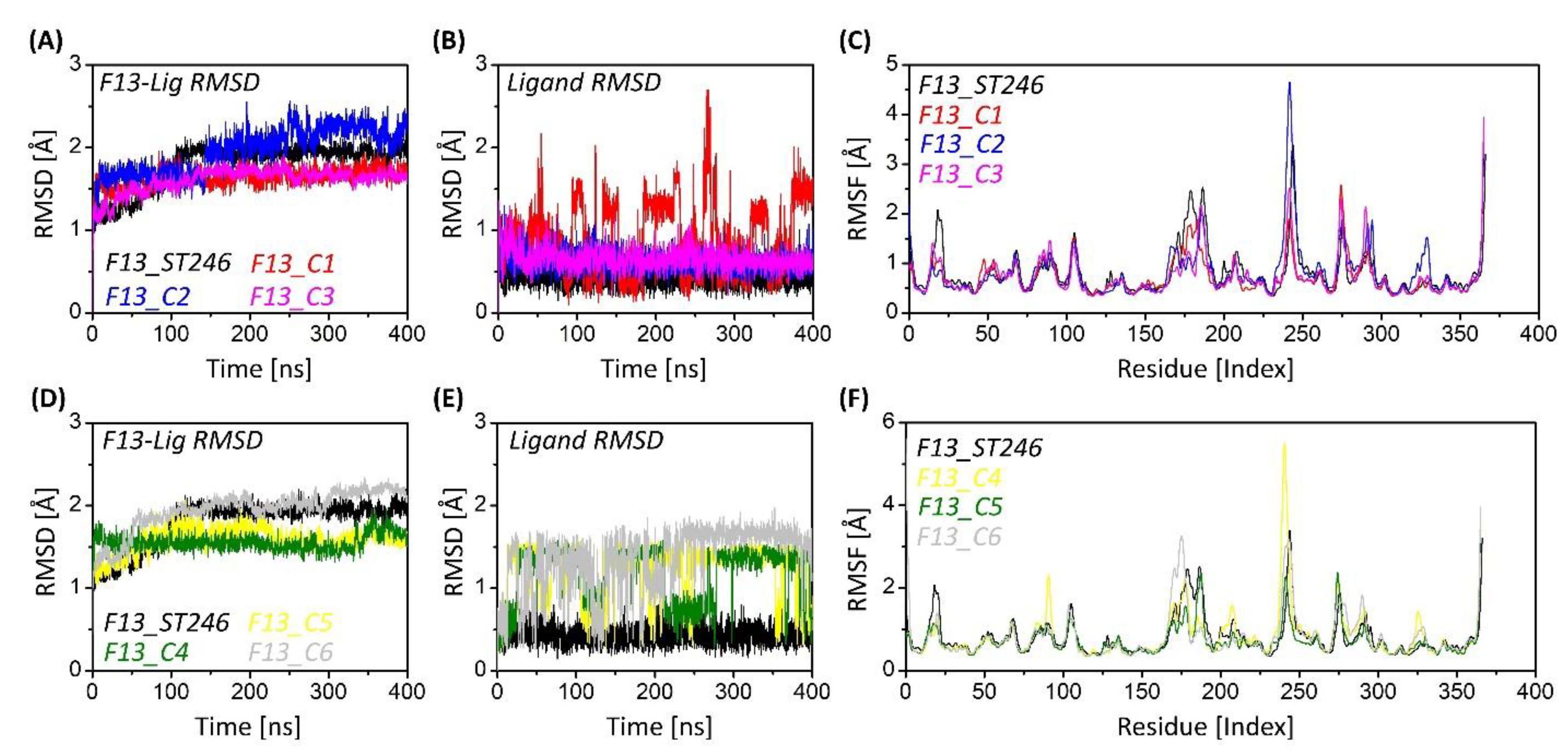
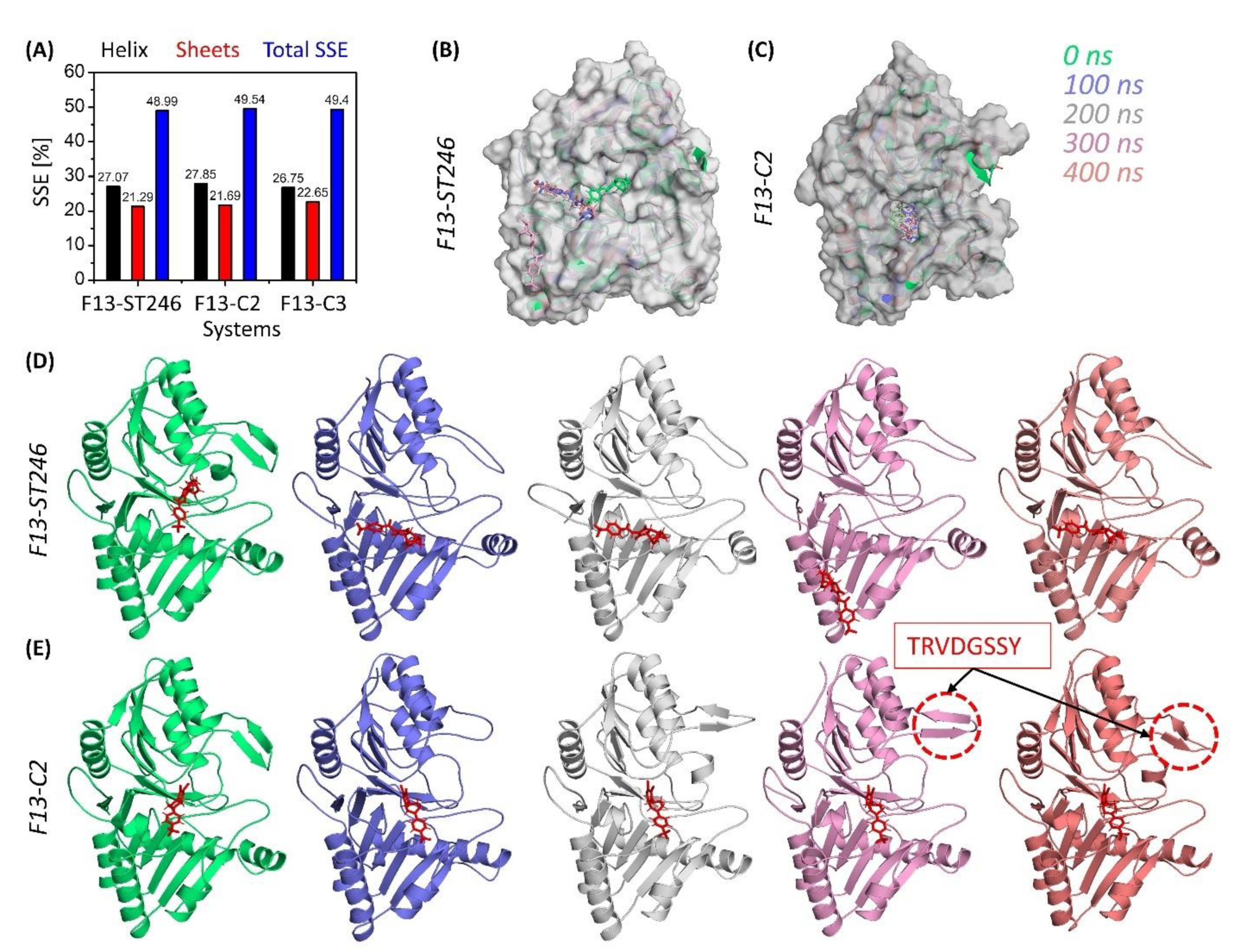
3.6. F13 Protein Stability upon Binding
3.7. F13-ST246 Complex and F13-C2 Complex Have Comparable Flexibility
3.8. F13-ST246 and F13-C2 Complexes Have Inline Solvent Accessibility and Compactness
3.9. F13 Protein in Complex with ST246 & C2 Enhance the Intraprotein hb Population
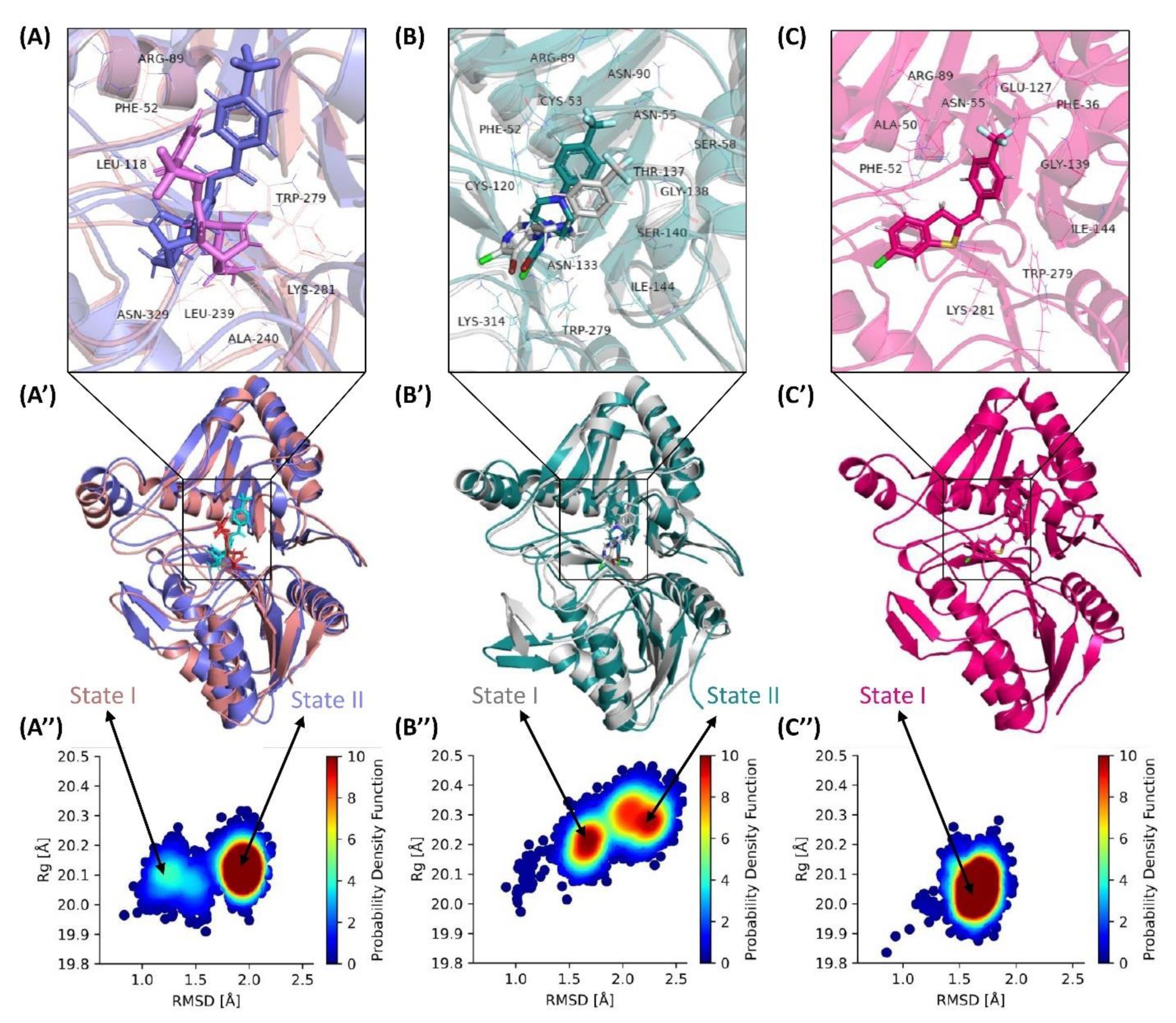
3.10. Exploring Transitional Conformations upon Binding of F13 Protein to ST246 & C2
3.11. C2 Binding to F13 Offer Conformational Stability
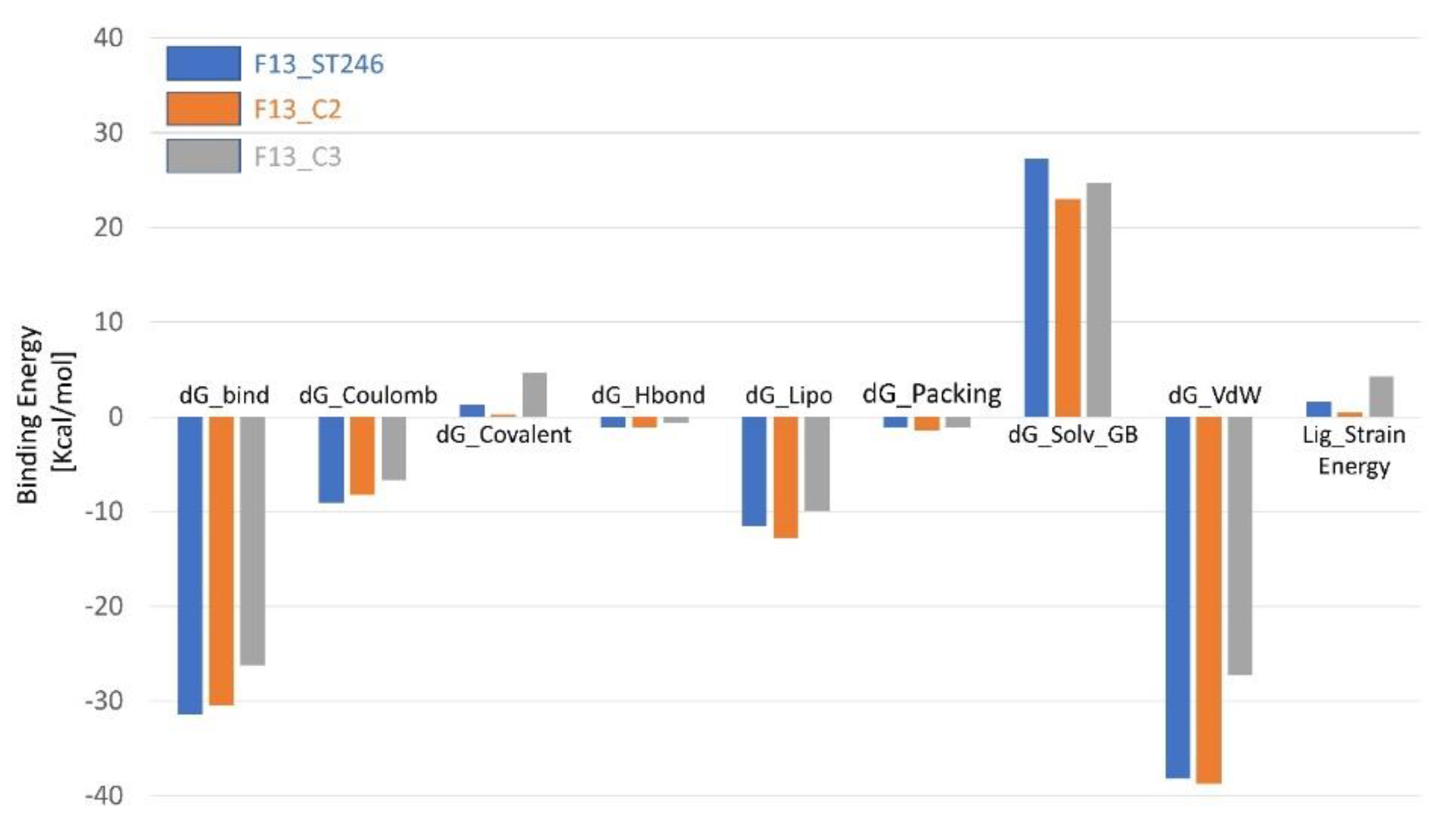
3.12. Binding Energy Calculation Shows a Favorable Profile for F13-C2 Complex
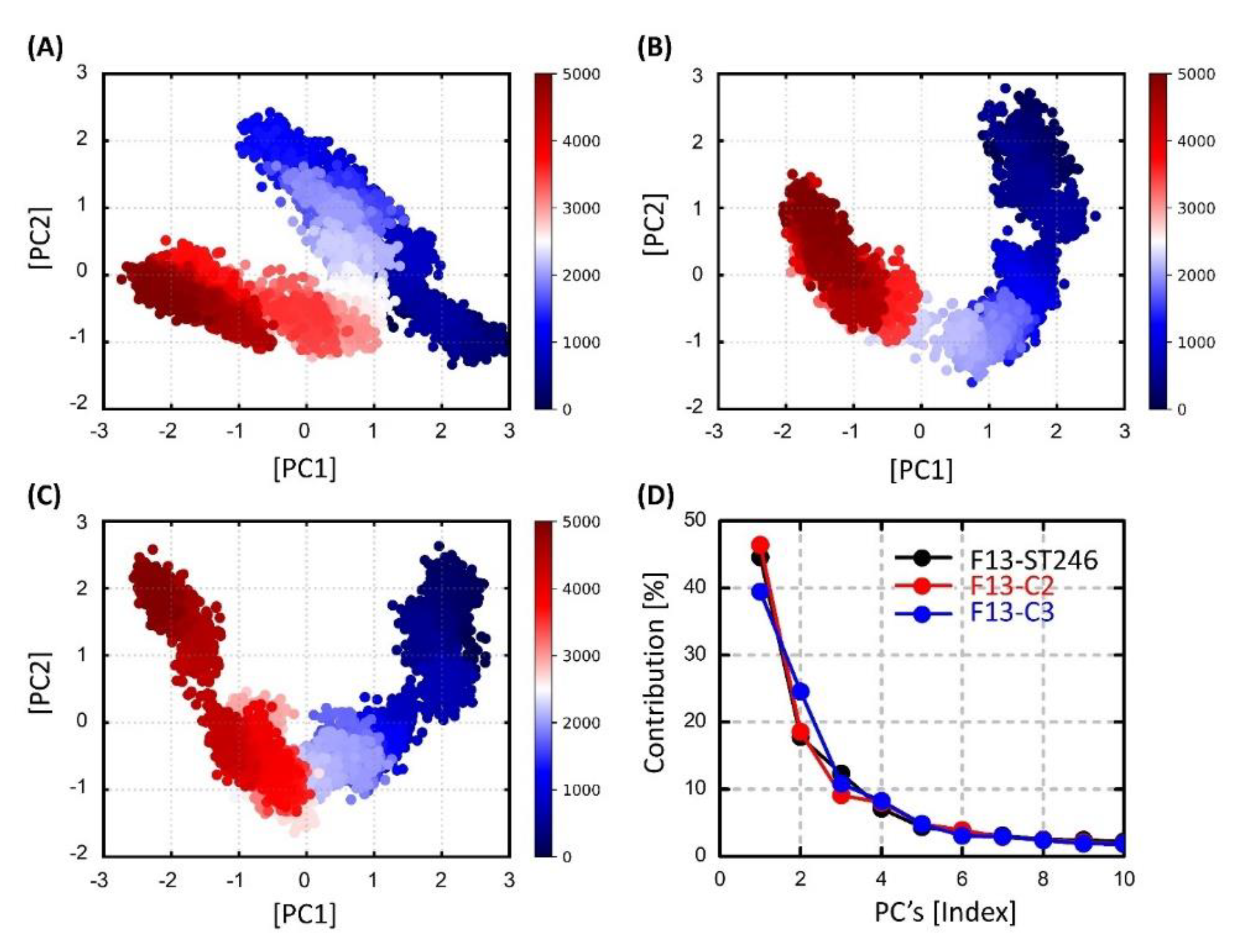
3.13. Dimensionality Reduction via Principal Components Analysis (PCA)
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- McCollum, A.M.; Damon, I.K. Human monkeypox. Clin. Infect. Dis. 2014, 58, 260–267. [Google Scholar] [CrossRef] [Green Version]
- WHO. Monkeypox Outbreak; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Antinori, A.; Mazzotta, V.; Vita, S.; Carletti, F.; Tacconi, D.; Lapini, L.E.; D’Abramo, A.; Cicalini, S.; Lapa, D.; Pittalis, S.; et al. Epidemiological, clinical and virological characteristics of four cases of monkeypox support transmission through sexual contact, Italy, May 2022. Eurosurveillance 2022, 27, 2200421. [Google Scholar] [CrossRef]
- Basgoz, N.; Brown, C.M.; Smole, S.C.; Madoff, L.C.; Biddinger, P.D.; Baugh, J.J.; Shenoy, E.S. Case 24-2022: A 31-Year-Old Man with Perianal and Penile Ulcers, Rectal Pain, and Rash. N. Engl. J. Med. 2022, 387, 547–556. [Google Scholar] [CrossRef]
- Girometti, N.; Byrne, R.; Bracchi, M.; Heskin, J.; McOwan, A.; Tittle, V.; Gedela, K.; Scott, C.; Patel, S.; Gohil, J.; et al. Demographic and clinical characteristics of confirmed human monkeypox virus cases in individuals attending a sexual health centre in London, UK: An observational analysis. Lancet Infect. Dis. 2022, 22, 1321–1328. [Google Scholar] [CrossRef]
- Khattak, S.; Qaisar, M.; Zaman, S.; Khan, T.A.; Ali, Y.; Wu, D.D.; Ji, X.Y. Monkeypox virus preparation in Pakistan-Next viral zoonotic disease outbreak after COVID-19? Biomed. Lett. 2022, 8, 196–201. [Google Scholar]
- Adler, H.; Gould, S.; Hine, P.; Snell, L.B.; Wong, W.; Houlihan, C.F.; Osborne, J.C.; Rampling, T.; Beadsworth, M.B.; Duncan, C.J.; et al. Clinical features and management of human monkeypox: A retrospective observational study in the UK. Lancet Infect. Dis. 2022, 22, 1153–1162. [Google Scholar] [CrossRef]
- De Baetselier, I.; Van Dijck, C.; Kenyon, C.; Coppens, J.; Michiels, J.; de Block, T.; Smet, H.; Coppens, S.; Vanroye, F.; Bugert, J.J.; et al. Retrospective detection of asymptomatic monkeypox virus infections among male sexual health clinic attendees in Belgium. Nat. Med. 2022, 28, 2288–2292. [Google Scholar] [CrossRef]
- Miura, F.; van Ewijk, C.E.; Backer, J.A.; Xiridou, M.; Franz, E.; de Coul, E.O.; Brandwagt, D.; van Cleef, B.; van Rijckevorsel, G.; Swaan, C.; et al. Estimated incubation period for monkeypox cases confirmed in the Netherlands. May Eurosurveillance 2022, 27, 2200448. [Google Scholar] [CrossRef]
- Huggins, J.; Goff, A.; Hensley, L.; Mucker, E.; Shamblin, J.; Wlazlowski, C.; Johnson, W.; Chapman, J.; Larsen, T.; Twenhafel, N.; et al. Nonhuman Primates Are Protected from Smallpox Virus or Monkeypox Virus Challenges by the Antiviral Drug ST-Antimicrob. Agents Chemother. 2009, 53, 2620–2625. [Google Scholar] [CrossRef] [Green Version]
- Jordan, R.; Goff, A.; Frimm, A.; Corrado, M.L.; Hensley, L.; Byrd, C.M.; Mucker, E.; Shamblin, J.; Bolken, T.C.; Wlazlowski, C.; et al. ST-246 Antiviral Efficacy in a Nonhuman Primate Monkeypox Model: Determination of the Minimal Effective Dose and Human Dose Justification. Antimicrob. Agents Chemother. 2009, 53, 1817–1822. [Google Scholar] [CrossRef] [Green Version]
- Grosenbach, D.W.; Honeychurch, K.; Rose, E.A.; Chinsangaram, J.; Frimm, A.; Maiti, B.; Lovejoy, C.; Meara, I.; Long, P.; Hruby, D.E. Oral Tecovirimat for the Treatment of Smallpox. N. Engl. J. Med. 2018, 379, 44–53. [Google Scholar] [CrossRef]
- Delaune, D.; Iseni, F. Drug Development against Smallpox: Present and Future. Antimicrob. Agents Chemother. 2020, 64, e01683-19. [Google Scholar] [CrossRef]
- EMA. Tecovirimat SIGA; SIGA Technologies, Inc.: Breda, The Netherlands, 2022. [Google Scholar]
- Yang, G.; Pevear, D.C.; Davies, M.H.; Collett, M.S.; Bailey, T.; Rippen, S.; Barone, L.; Burns, C.; Rhodes, G.; Tohan, S.; et al. An Orally Bioavailable Antipoxvirus Compound (ST-246) Inhibits Extracellular Virus Formation and Protects Mice from Lethal Orthopoxvirus Challenge. J. Virol. 2005, 79, 13139–13149. [Google Scholar] [CrossRef] [Green Version]
- Payne, L.G. Significance of Extracellular Enveloped Virus in the in vitro and in vivo Dissemination of Vaccinia. J. Gen. Virol. 1980, 50, 89–100. [Google Scholar] [CrossRef]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 2002, 83, 2915–2931. [Google Scholar] [CrossRef]
- Russo, A.T.; Grosenbach, D.W.; Chinsangaram, J.; Honeychurch, K.M.; Long, P.G.; Lovejoy, C.; Maiti, B.; Meara, I.; Hruby, D.E. An overview of tecovirimat for smallpox treatment and expanded anti-orthopoxvirus applications. Expert Rev. Anti-infective Ther. 2021, 19, 331–344. [Google Scholar] [CrossRef]
- Frenois-Veyrat, G.; Gallardo, F.; Gorgé, O.; Marcheteau, E.; Ferraris, O.; Baidaliuk, A.; Favier, A.L.; Enfroy, C.; Holy, X.; Lourenco, J.; et al. Tecovirimat is effective against human monkeypox virus in vitro at nanomolar concentrations. Nat. Microbiol. 2022, 7, 1951–1955. [Google Scholar] [CrossRef]
- Khattak, S.; Rauf, M.A.; Zaman, Q.; Ali, Y.; Fatima, S.; Muhammad, P.; Li, T.; Khan, H.A.; Khan, A.A.; Ngowi, E.E.; et al. Genome-Wide Analysis of Codon Usage Patterns of SARS-CoV-2 Virus Reveals Global Heterogeneity of COVID. Biomolecules 2021, 11, 912. [Google Scholar] [CrossRef]
- Seib, K.; Dougan, G.; Rappuoli, R. The Key Role of Genomics in Modern Vaccine and Drug Design for Emerging Infectious Diseases. PLoS Genet. 2009, 5, e1000612. [Google Scholar] [CrossRef] [Green Version]
- Sahoo, B.M.; Kumar, B.V.V.R.; Sruti, J.; Mahapatra, M.K.; Banik, B.K.; Borah, P. Drug Repurposing Strategy (DRS): Emerging Approach to Identify Potential Therapeutics for Treatment of Novel Coronavirus Infection. Front. Mol. Biosci. 2021, 8, 628144. [Google Scholar] [CrossRef]
- Choudhury, C.; Murugan, N.A.; Priyakumar, U.D. Structure-based drug repurposing: Traditional and advanced AI/ML-aided methods. Drug Discov. Today 2022, 27, 1847–1861. [Google Scholar] [CrossRef]
- Ajmal, A.; Ali, Y.; Khan, A.; Wadood, A.; Rehman, A.U. Identification of novel peptide inhibitors for the KRas-G12C variant to prevent oncogenic signaling. Biomol. Struct. Dyn. 2022, 40, 1–10. [Google Scholar] [CrossRef]
- Murray, C.W.; Blundell, T.L. Structural biology in fragment-based drug design. Curr. Opin. Struct. Biol. 2010, 20, 497–507. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Tunyasuvunakool, K.; Ronneberger, O.; Bates, R.; Žídek, A.; Bridgland, A.; et al. AlphaFold 2. In Fourteenth Critical Assessment of Techniques for Protein Structure Prediction; DeepMind: London, UK, 2020. [Google Scholar]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef] [Green Version]
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Colovos, C.; Yeates, T.O. Verification of protein structures: Patterns of nonbonded atomic interactions. Protein Sci. 1993, 2, 1511–1519. [Google Scholar] [CrossRef] [Green Version]
- Shan, J.; Pan, X.; Wang, X.; Xiao, X.; Ji, C. FragRep: A Web Server for Structure-Based Drug Design by Fragment Replacement. J. Chem. Inf. Model. 2020, 60, 5900–5906. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Li, K.; Zhang, L. Targeting F13 from monkeypox virus and variola virus by tecovirimat: Molecular simulation analysis. J. Infect. 2022, 85, e99–e101. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Accelrys Ltd. , Discovery Studio, v2.1; Accelrys Ltd.: San Diego, CA, USA, 2008. [Google Scholar]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006. [Google Scholar]
- Ali, Y.; Ahmad, F.; Ullah, M.F.; Haq, N.U.; Haq, M.I.U.; Aziz, A.; Zouidi, F.; Khan, M.I.; Eldin, S.M. Structural Evaluation and Conformational Dynamics of ZNF141T474I Mutation Provoking Postaxial Polydactyly Type A. Bioengineering 2022, 9, 749. [Google Scholar] [CrossRef]
- Shah, A.A.; Amjad, M.; Hassan, J.-U.; Ullah, A.; Mahmood, A.; Deng, H.; Ali, Y.; Gul, F.; Xia, K. Molecular Insights into the Role of Pathogenic nsSNPs in GRIN2B Gene Provoking Neurodevelopmental Disorders. Genes 2022, 13, 1332. [Google Scholar] [CrossRef]
- Ahmad, S.U.; Ali, Y.; Jan, Z.; Rasheed, S.; Nazir, N.U.A.; Khan, A.; Abbas, S.R.; Wadood, A.; Rehman, A.U. Computational screening and analysis of deleterious nsSNPs in human p14ARF (CDKN2A gene) protein using molecular dynamic simulation approach. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Kräutler, V.; Van Gunsteren, W.F.; Hünenberger, P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001, 22, 501–508. [Google Scholar] [CrossRef]
- Shaw, D.E. A fast, scalable method for the parallel evaluation of distance-limited pairwise particle interactions. J. Comput. Chem. 2005, 26, 1318–1328. [Google Scholar] [CrossRef] [Green Version]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar] [CrossRef] [Green Version]
- Martyna, G.J.; Tuckerman, M.E.; Tobias, D.J.; Klein, M.L. Explicit reversible integrators for extended systems dynamics. Mol. Phys. 1996, 87, 1117–1157. [Google Scholar] [CrossRef]
- Bakan, A.; Meireles, L.M.; Bahar, I. ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [Google Scholar] [CrossRef] [Green Version]
- Harris, C.R.; Millman, K.J.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins: Struct. Funct. Bioinform. 2011, 79, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Galzitskaya, O.V.; Garbuzynskiy, S.O. Entropy capacity determines protein folding. Proteins: Struct. Funct. Bioinform. 2006, 63, 144–154. [Google Scholar] [CrossRef]
- Abresch, E.C.; Paddock, M.L.; Villalobos, M.; Chang, C.; Okamura, M.Y. Interaction between Cytochrome c2 and the Photosynthetic Reaction Center from Rhodobacter sphaeroides: Role of Interprotein Hydrogen Bonds in Binding and Electron Transfer. Biochemistry 2008, 47, 13318–13325. [Google Scholar] [CrossRef] [Green Version]
- Hubbard, R.E.; Kamran Haider, M. Hydrogen Bonds in Proteins: Role and Strength. eLS 2010. [Google Scholar] [CrossRef]
- Ladnyj, I.D.; Ziegler, P.; Kima, E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull. World Heal. Organ. 1972, 46, 593–597. [Google Scholar]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A tale of two clades: Monkeypox viruses. J. Gen. Virol. 2005, 86, 2661–2672. [Google Scholar] [CrossRef]
- Reed, K.D.; Melski, J.W.; Graham, M.B.; Regnery, R.L.; Sotir, M.J.; Wegner, M.V.; Kazmierczak, J.J.; Stratman, E.J.; Li, Y.; Fairley, J.A.; et al. The Detection of Monkeypox in Humans in the Western Hemisphere. N. Engl. J. Med. 2004, 350, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Khattak, S.; Rauf, M.A.; Ali, Y.; Yousaf, M.T.; Liu, Z.; Wu, D.D.; Ji, X.Y. The monkeypox diagnosis, treatments and prevention: A review. Front. Cell. Infect. Microbiol. 2023, 12, 2005. [Google Scholar] [CrossRef]
- Minasov, G.; Inniss, N.L.; Shuvalova, L.; Anderson, W.F.; Satchell, K.J.F. Structure of the Monkeypox virus profilin-like protein A42R reveals potential functional differences from cellular profilins. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2022, 78, 371–377. [Google Scholar] [CrossRef]
- Moss, B. Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef]
- Bryk, P.; Brewer, M.G.; Ward, B.M. Vaccinia Virus Phospholipase Protein F13 Promotes Rapid Entry of Extracellular Virions into Cells. J. Virol. 2018, 92, e02154-17. [Google Scholar] [CrossRef] [Green Version]
- Rehman, A.U.; Khurshid, B.; Ali, Y.; Rasheed, S.; Wadood, A.; Ng, H.-L.; Chen, H.-F.; Wei, Z.; Luo, R.; Zhang, J. Computational approaches for the design of modulators targeting protein-protein interactions. Expert Opin. Drug Discov. 2023. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Property | Value | Property | Value |
|---|---|---|---|
| Number of atoms | 27 | Lipinski Hyd. bond donor | 1 |
| Number of heteroatoms | 8 | TPSA | 66.48 |
| HBA | 3 | Lipinski Hyd. bond acceptors | 5 |
| Aromatic Carbocycles | 1 | Rings | 6 |
| LogP | 2.403 | Mol Wt | 372.103 |
| RB | 2 | Labute ASA | 151.724 |
| Aromatic Heterocycles | 0 | Aromatic rings | 1 |
| No. of Aliphatic Carbocycles | 4 |
| Compound | Binding Energy (Kcal/mol) | Hydrogen Bonds | Hydrophobic Interactions |
|---|---|---|---|
| ST-246 | −8.3 | Asn55, Arg89, Ser58 | Asn55 Arg89, Cys53, Phe52, Leu118 |
| C1 | −8.9 | Arg89, Asn55 | Phe52, Lys281, Cys53, Arg89, |
| C2 | −9.2 | Arg89, Asn55, Asn312, Thr137, Ser135, Ser327 | Cys53, Leu239, Lys314, Cys53 |
| C3 | −7.03 | Arg89, Thr137, Lys281 | Cys53, Arg89, Lys281 |
| C4 | −7.1 | Ser58 | Cys53, Arg89, Phe52 |
| C5 | −6.9 | Asn55 | Cys53, Phe52, Arg89, Leu118, Lys281 |
| C6 | −6.6 | Cys53, Arg89 | Leu118, Phe52 |
| Compounds | MW | RB | HBA | HBD | TPSA | Log P |
|---|---|---|---|---|---|---|
| ST-246 | 376.33 | 4 | 6 | 1 | 66.48 | 2.65 |
| C1 | 328.78 | 3 | 3 | 0 | 25.3 | 5.64 |
| C2 | 421.64 | 3 | 5 | 0 | 32.26 | 3.94 |
| C3 | 317.35 | 4 | 3 | 0 | 4.41 | 5.47 |
| C4 | 392.45 | 3 | 3 | 1 | 72.96 | 1.38 |
| C5 | 410.42 | 3 | 5 | 1 | 92.26 | 2.23 |
| C6 | 397.85 | 4 | 3 | 1 | 69.72 | 2.17 |
| Compounds | Solubility | BBB Permeant | Pharmacokinetics | |||
|---|---|---|---|---|---|---|
| Log S (ESOL) | Log S (Ali) | Log S (SILICOS-IT) | GI Absorption | CYP Enzymes Inhibitors | ||
| ST-246 | Soluble | Soluble | Soluble | Yes | High | CYP2C19 and CYP3A4 inhibitor |
| C1 | Moderately soluble | Poorly soluble | Poorly soluble | No | Low | CYP1A2, CYP2C9, and CYP2D6 inhibitor |
| C2 | Moderately soluble | Moderately soluble | Moderately soluble | Yes | High | CYP1A2, CYP2C19, and CYP2C9 inhibitor |
| C3 | Poorly soluble | Poorly soluble | Poorly soluble | No | Low | CYP1A2, CYP2C19, and CYP2D6 inhibitor |
| C4 | Soluble | Soluble | Soluble | No | High | CYP3A4 inhibitor |
| C5 | Moderately soluble | Moderately soluble | Moderately soluble | No | High | CYP1A2 inhibitor |
| C6 | Soluble | Soluble | Soluble | No | High | CYP2C19, CYP2D6, and CYP3A4 inhibitor |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ali, Y.; Imtiaz, H.; Tahir, M.M.; Gul, F.; Saddozai, U.A.K.; ur Rehman, A.; Ren, Z.-G.; Khattak, S.; Ji, X.-Y. Fragment-Based Approaches Identified Tecovirimat-Competitive Novel Drug Candidate for Targeting the F13 Protein of the Monkeypox Virus. Viruses 2023, 15, 570. https://doi.org/10.3390/v15020570
Ali Y, Imtiaz H, Tahir MM, Gul F, Saddozai UAK, ur Rehman A, Ren Z-G, Khattak S, Ji X-Y. Fragment-Based Approaches Identified Tecovirimat-Competitive Novel Drug Candidate for Targeting the F13 Protein of the Monkeypox Virus. Viruses. 2023; 15(2):570. https://doi.org/10.3390/v15020570
Chicago/Turabian StyleAli, Yasir, Hina Imtiaz, Muhammad Mutaal Tahir, Fouzia Gul, Umair Ali Khan Saddozai, Ashfaq ur Rehman, Zhi-Guang Ren, Saadullah Khattak, and Xin-Ying Ji. 2023. "Fragment-Based Approaches Identified Tecovirimat-Competitive Novel Drug Candidate for Targeting the F13 Protein of the Monkeypox Virus" Viruses 15, no. 2: 570. https://doi.org/10.3390/v15020570