
Genomic Epidemiology and Transmission Dynamics of Global Coxsackievirus B4

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Virus Isolation and Ethical Considerations
2.2. RNA Extraction, Library Construction, and NGS
2.3. CVB4 Dataset Construction
2.4. Phylogenetic and Recombination Analysis
2.5. Amino Acid Site Variation Analysis
2.6. Temporal Dynamics Analysis
2.7. Geographical Clustering Intensity and Phylogeographic Analysis
3. Results
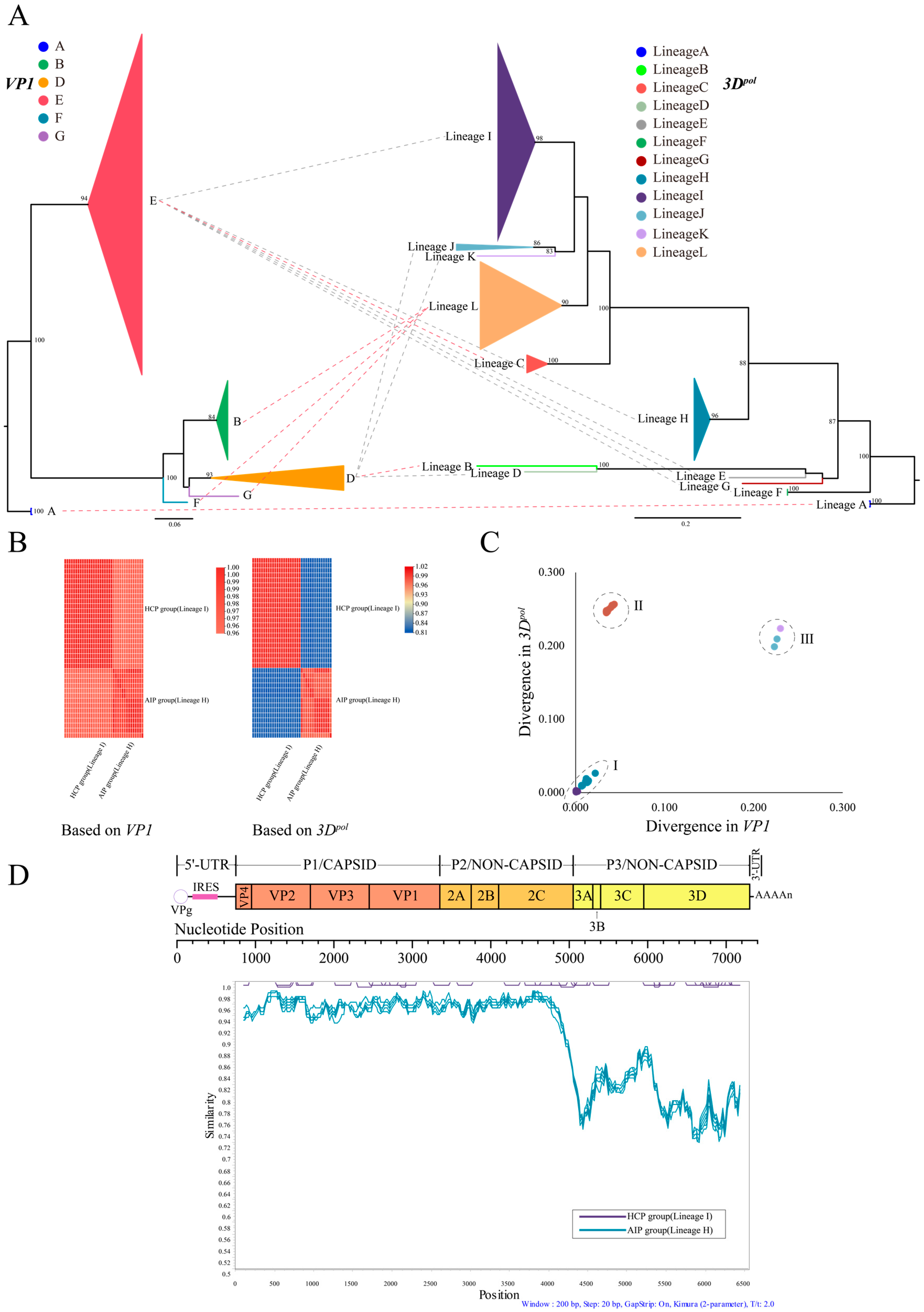
3.1. Phylogenetic Analysis Based on CVB4 Whole-Genome Sequences
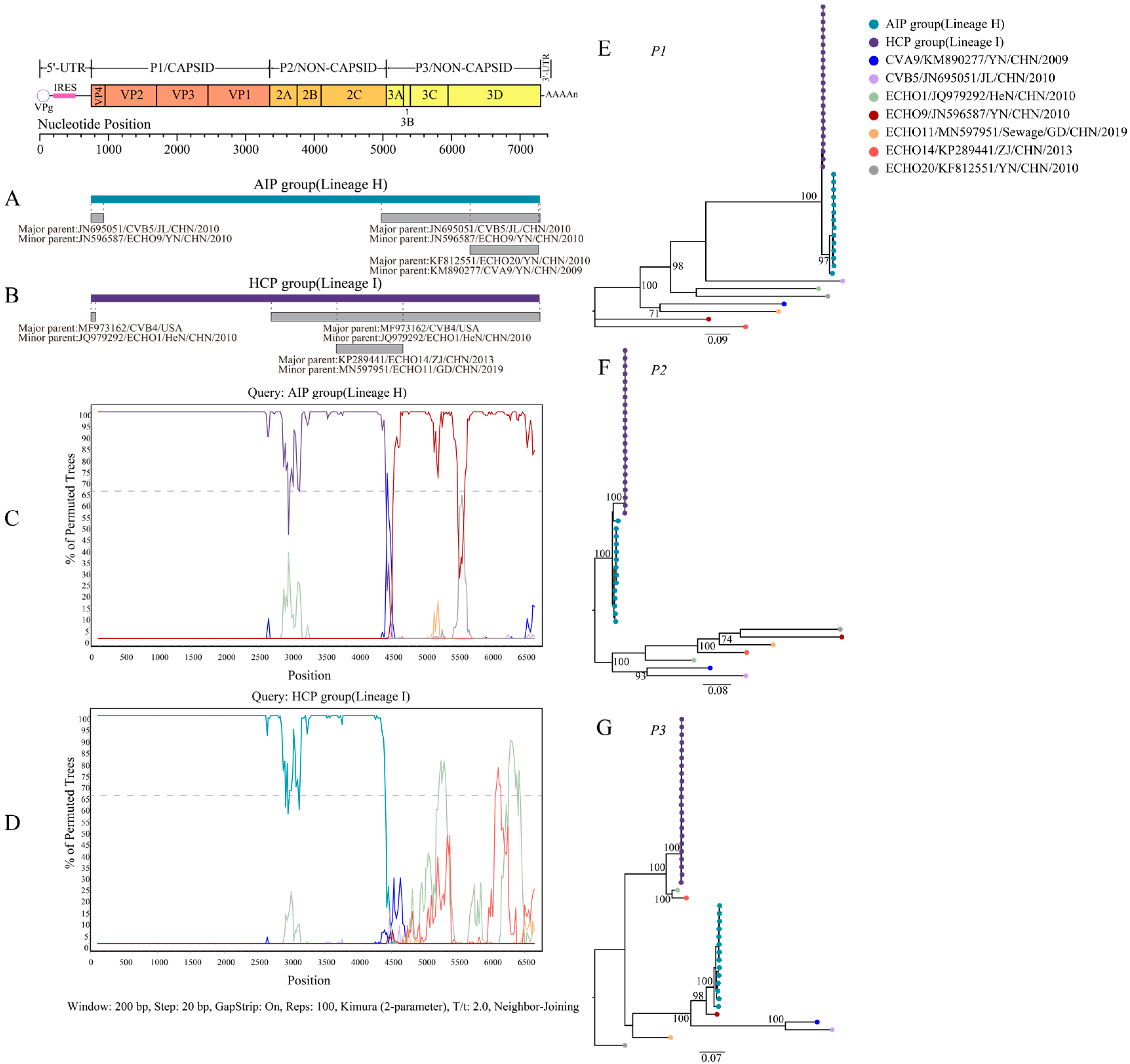
3.2. Recombination of CVB4 with Other Enteroviruses
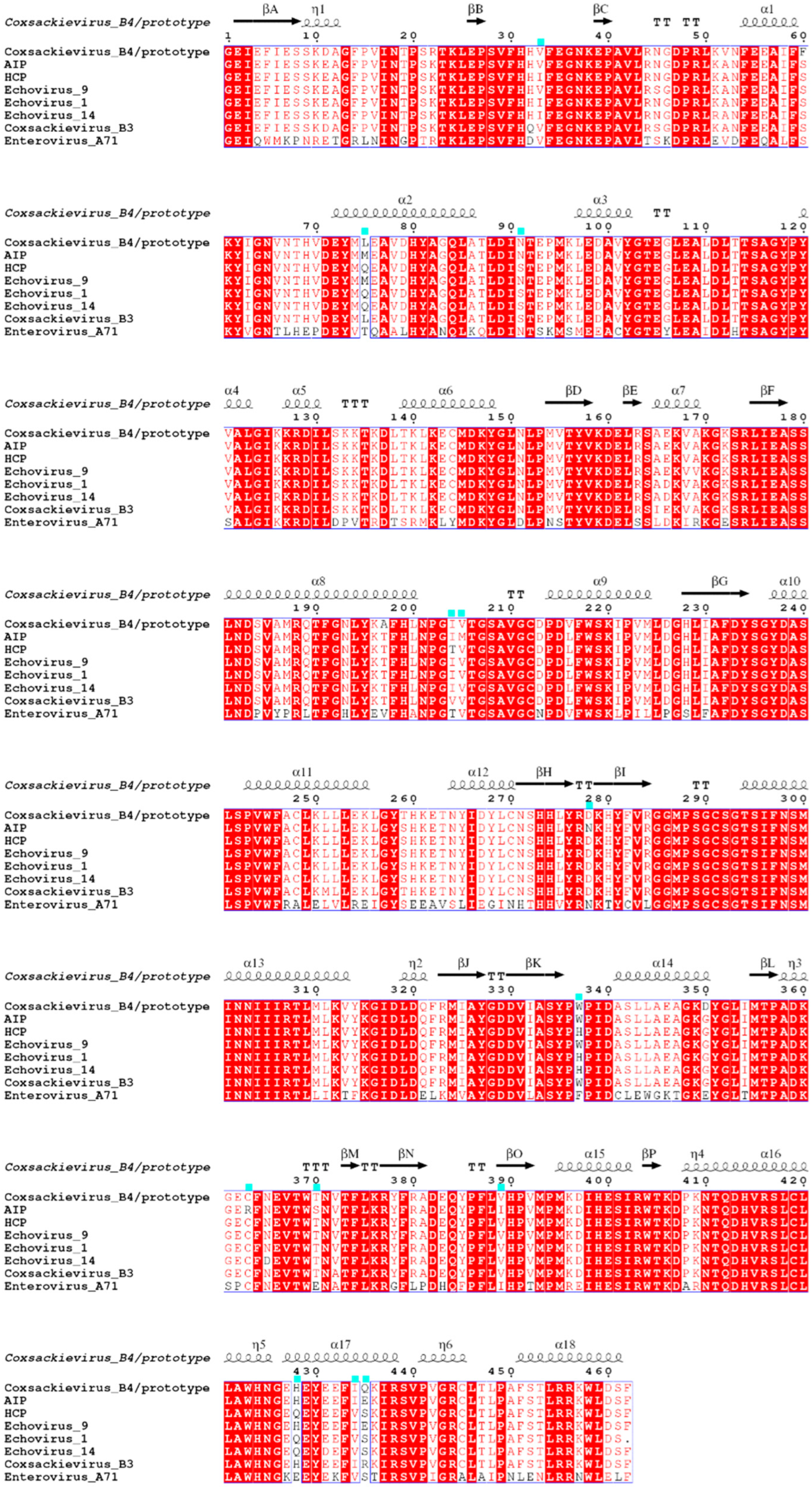
3.3. Variation Characteristics of CVB4 Amino Acid Position in the P3 Region
3.4. Evolutionary Dynamics of CVB4
3.5. Phylogeographic Analysis of CVB4 Dominant Genotypes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Knowles, N.J.; Hovi, T.; King, A.M.Q.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Simmonds, P.; Skern, T.; Stanway, G.; Yamashita, T.; et al. Picornaviridae. In Virus Taxonomy: Classification and Nomenclature of Viruses: Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: San Diego, CA, USA, 2012; pp. 855–880. [Google Scholar]
- Han, Z.; Zhang, Y.; Huang, K.; Wang, J.; Tian, H.; Song, Y.; Yang, Q.; Yan, D.; Zhu, S.; Yao, M.; et al. Two Coxsackievirus B3 outbreaks associated with hand, foot, and mouth disease in China and the evolutionary history worldwide. BMC Infect Dis. 2019, 19, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, N.; Jia, L.; Yin, J.; Wu, Z.; Wang, Z.; Li, P.; Hao, R.; Wang, L.; Wang, Y.; Qiu, S.; et al. An outbreak of aseptic meningitis caused by a distinct lineage of coxsackievirus B5 in China [Research Support, Non-U.S. Gov’t]. Int. J. Infect Dis. 2014, 23, 101–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, X.; Zhang, Y.; Gu, S.; Fan, Y.; Sun, Q.; Zhang, B.; Yan, S.; Xu, W.; Ma, X.; Wang, W. New coxsackievirus B4 genotype circulating in Inner Mongolia Autonomous Region, China. PLoS ONE 2014, 9, e90379. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Li, Y.; Liu, G.; Jiang, Y.; Shen, S.; Bi, R.; Huang, H.; Cheng, T.; Wang, C.; Wei, W. A second open reading frame in human enterovirus determines viral replication in intestinal epithelial cells. Nat. Commun. 2019, 10, 4066. [Google Scholar] [CrossRef] [Green Version]
- Oberste, M.S.; Maher, K.; Williams, A.J.; Dybdahl-Sissoko, N.; Brown, B.A.; Gookin, M.S.; Penaranda, S.; Mishrik, N.; Uddin, M.; Pallansch, M.A. Species-specific RT-PCR amplification of human enteroviruses: A tool for rapid species identification of uncharacterized enteroviruses. J. Gen. Virol. 2006, 87, 119–128. [Google Scholar] [CrossRef]
- Lindberg, A.M.; Andersson, P.; Savolainen, C.; Mulders, M.N.; Hovi, T. Evolution of the genome of Human enterovirus B: Incongruence between phylogenies of the VP1 and 3CD regions indicates frequent recombination within the species. J. Gen. Virol. 2003, 84, 1223–1235. [Google Scholar] [CrossRef]
- Lu, J.; Kang, M.; Zeng, H.; Zhong, Y.; Fang, L.; Zheng, X.; Liu, L.; Yi, L.; Lin, H.; Peng, J.; et al. Tracking echovirus eleven outbreaks in Guangdong, China: A metatranscriptomic, phylogenetic, and epidemiological study. Virus Evol. 2020, 6, veaa029. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Q.; Li, J.; Cao, W.; Zhang, J.X.; Zhang, L.; Zhang, W.; Shao, Z.J.; Yan, Y. Analysis of recombination and natural selection in human enterovirus 71. Virology 2010, 398, 251–261. [Google Scholar] [CrossRef]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating Human enterovirus B: Independent evolution of structural and non-structural genome regions. J. Gen. Virol. 2005, 86, 3281–3290. [Google Scholar] [CrossRef]
- Xiao, J.; Wang, J.; Zhang, Y.; Sun, D.; Lu, H.; Han, Z.; Song, Y.; Yan, D.; Zhu, S.; Pei, Y.; et al. Coxsackievirus B4, an underestimated pathogen associated with a hand, foot, and mouth disease outbreak. Arch. Virol. 2021, 166, 2225–2234. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Han, T.; Tan, X.; Zhu, S.; Yan, D.; Yang, Q.; Song, Y.; Cui, A.; Zhang, Y.; Mao, N.; et al. Surveillance, epidemiology, and pathogen spectrum of hand, foot, and mouth disease in mainland of China from 2008 to 2017. Biosaf. Health 2019, 1, 32–40. [Google Scholar] [CrossRef]
- Xu, W.; Zhang, Y. Isolation and Characterization of Vaccine-Derived Polioviruses, Relevance for the Global Polio Eradication Initiative. Methods Mol. Biol. 2016, 1387, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Edgar. R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8, A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [Green Version]
- Goss, E.M.; Tabima, J.F.; Cooke, D.E.; Restrepo, S.; Fry, W.E.; Forbes, G.A.; Fieland, V.J.; Cardenas, M.; Grünwald, N.J. The Irish potato famine pathogen Phytophthora infestans originated in central Mexico rather than the Andes. Proc. Natl. Acad. Sci. USA 2014, 111, 8791–8796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11, Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Salminen, M.O.; Carr, J.K.; Burke, D.S.; McCutchan, F.E. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res. Hum. Retrovir. 1995, 11, 1423–1425. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4, Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Tan, Y.; Lam, T.T.; Heberlein-Larson, L.A.; Smole, S.C.; Auguste, A.J.; Hennigan, S.; Halpin, R.A.; Fedorova, N.; Puri, V.; Stockwell, T.B.; et al. Large-Scale Complete-Genome Sequencing and Phylodynamic Analysis of Eastern Equine Encephalitis Virus Reveals Source-Sink Transmission Dynamics in the United States. J. Virol. 2018, 92, e00074-18. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Kosakovsky Pond, S.L.; Scheffler, K. FUBAR: A fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieux, A.; Khatchikian, C.E. tipdatingbeast: An r package to assist the implementation of phylogenetic tip-dating tests using beast. Mol. Ecol. Resour. 2017, 17, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, J.; Rambaut, A.; Pybus, O.G. Correlating viral phenotypes with phylogeny: Accounting for phylogenetic uncertainty. Infect. Genet. Evol. 2008, 8, 239–246. [Google Scholar] [CrossRef]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian phylogeography finds its roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Rambaut, A.; Suchard, M.A.; Lemey, P. SPREAD: Spatial phylogenetic reconstruction of evolutionary dynamics. Bioinformatics 2011, 27, 2910–2912. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Liu, X.; Du, Z.; Hou, H.; Wang, X.; Wang, F.; Yang, J. Bayesian phylodynamic analysis reveals the dispersal patterns of tobacco mosaic virus in China. Virology 2019, 528, 110–117. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2013, 6, 1110–1116. [Google Scholar] [CrossRef]
- Bandelt, H.J.; Forster, P.; Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Mulders, M.N.; Salminen, M.; Kalkkinen, N.; Hovi, T. Molecular epidemiology of coxsackievirus B4 and disclosure of the correct VP1/2A(pro) cleavage site: Evidence for high genomic diversity and long-term endemicity of distinct genotypes. J. Gen. Virol. 2000, 81, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Song, Y.; Xiao, J.; Jiang, L.; Huang, W.; Wei, H.; Li, J.; Zeng, H.; Yu, Q.; Li, J.; et al. Genomic epidemiology of coxsackievirus A16 in mainland of China, 2000–2018. Virus Evol. 2020, 6, veaa084. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.C.; Ananda Babu, M.; Raghavendra, A.; Dhananjaya, D.; Kumar, S.; Maiya, P.P. Non-polio enteroviruses and their association with acute diarrhea in children in India. Infect Genet. Evol. 2013, 17, 153–161. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method of Analysis and Dataset | Mean dN/dS | No. of Negative/Purifying Selection Sites | Position(s) of Positive/Diversifying Selection Site (codon) |
|---|---|---|---|
| SLAC a | |||
| HCP group | 0.0161 | 2 | |
| AIP group | 0.0258 | 6 | |
| FUBAR b | |||
| HCP group | 416 | 347 | |
| AIP group | 408 |
| Lineage | Genotype | N a | Divergence b | Isolated Countries | Isolated Years |
|---|---|---|---|---|---|
| A | A | 2 | 0.002 | The United States, Italy | 1951 |
| B | D | 1 | NA c | Romania | 1986 |
| C | E | 4 | 0.027 | Australia, China | 2007–2008 |
| D | D | 1 | NA | Cameroon | 2008 |
| E | E | 1 | NA | China | 2009 |
| F | E | 2 | 0.000 | Australia | 2010 |
| G | E | 1 | NA | China | 2010 |
| H | E | 15 | 0.019 | China | 2010, 2013 |
| I | E | 34 | 0.041 | China | 2010–2014 |
| J | D | 2 | 0.193 | Australia, France | 2011, 2019 |
| K | D | 1 | NA | The United States | 2016 |
| L | B, F, G | 16 | 0.105 | Denmark | NA d |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, J.; Wang, J.; Lu, H.; Song, Y.; Sun, D.; Han, Z.; Li, J.; Yang, Q.; Yan, D.; Zhu, S.; et al. Genomic Epidemiology and Transmission Dynamics of Global Coxsackievirus B4. Viruses 2023, 15, 569. https://doi.org/10.3390/v15020569
Xiao J, Wang J, Lu H, Song Y, Sun D, Han Z, Li J, Yang Q, Yan D, Zhu S, et al. Genomic Epidemiology and Transmission Dynamics of Global Coxsackievirus B4. Viruses. 2023; 15(2):569. https://doi.org/10.3390/v15020569
Chicago/Turabian StyleXiao, Jinbo, Jianxing Wang, Huanhuan Lu, Yang Song, Dapeng Sun, Zhenzhi Han, Jichen Li, Qian Yang, Dongmei Yan, Shuangli Zhu, and et al. 2023. "Genomic Epidemiology and Transmission Dynamics of Global Coxsackievirus B4" Viruses 15, no. 2: 569. https://doi.org/10.3390/v15020569