
SARS-CoV-2 Omicron Subvariants Balance Host Cell Membrane, Receptor, and Antibody Docking via an Overlapping Target Site

Abstract
:1. Introduction
2. Materials and Methods
2.1. Spike Sequence Analysis
2.2. Structures of Spike Proteins
2.3. Membrane Binding Sites
2.4. Statistical Analysis
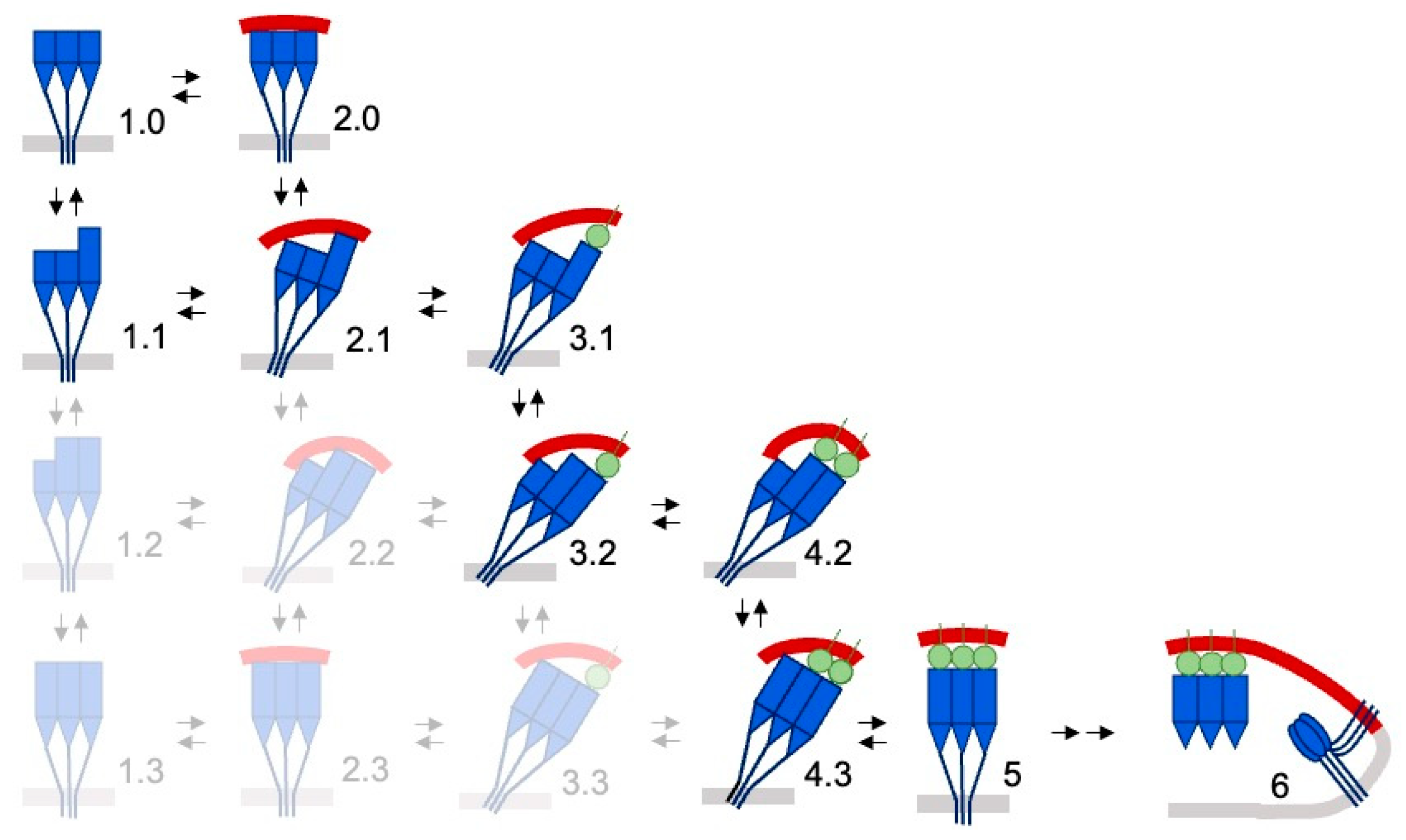
3. Results
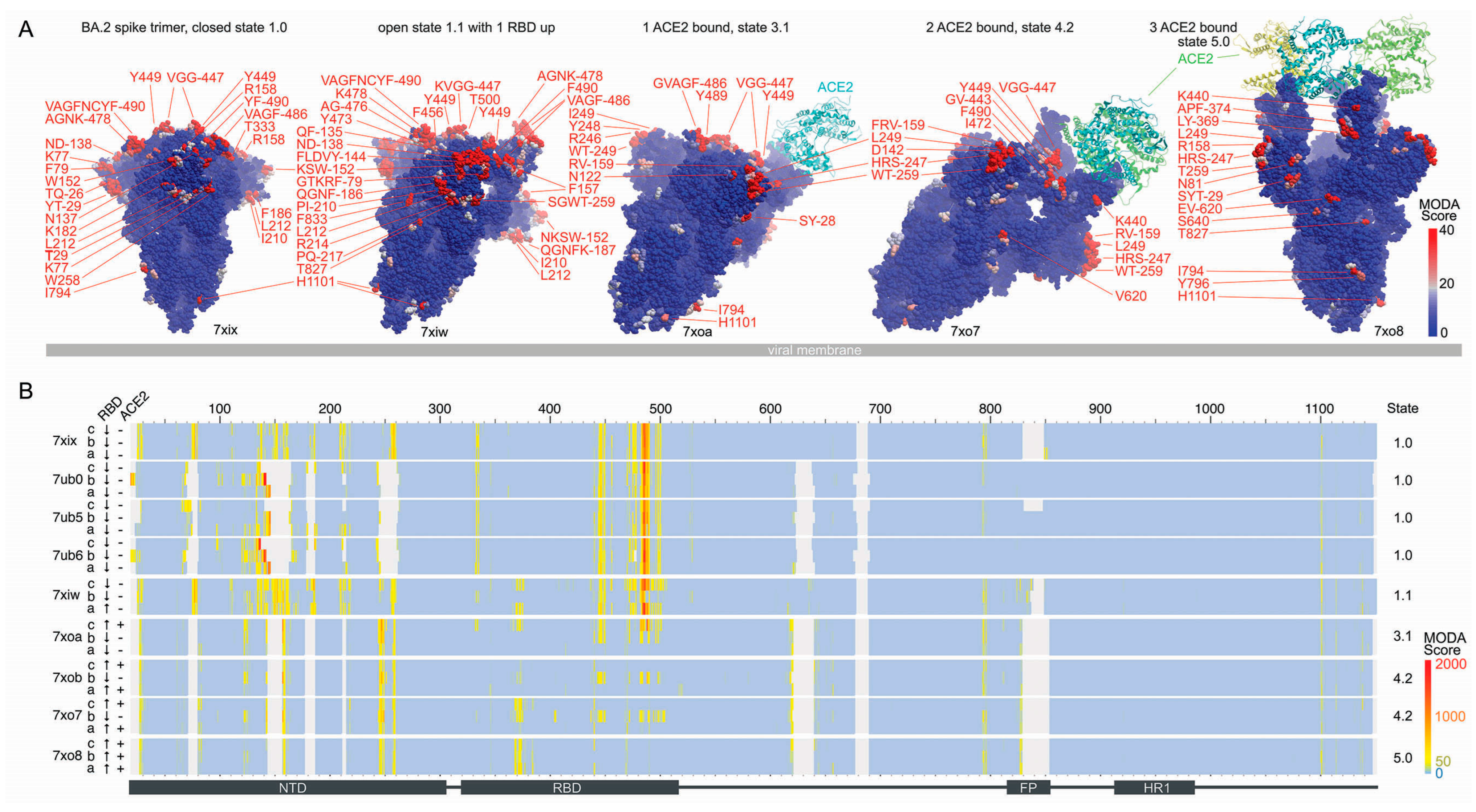
3.1. Conformations and Membrane Binding of BA.2 Spike Trimers
- The T19I mutation in BA.2 adds exposed hydrophobicity and increases the membrane binding score of this position from 33.8 ± 10.8 to 214.1 in structures where this element is best resolved (PDB: 7tnw vs. 7ub0-b).
- The gain of the ΔL24, ΔP25, ΔP26, and A27S mutations in BA.2 increases the degree of disorder locally, although the S27 residue is resolved with significant membrane binding propensities in all the BA.2 spike:ACE-2 complexes (PDBs 7xoa, 7xob, 7xo7, 7xo8, Figure 2).
- The loss of the A67V, ΔH69, and ΔV70 mutations in BA2 leads to significant membrane binding propensities appearing at the H69 and V70 positions in structures where this loop is resolved (PDB: 7ub5).
- The loss of the T95I mutation in BA2 eliminates the membrane binding propensity that appears at this position in a minority of structures, such as the nearby K97. Still, it occasionally appears as membrane-binding in closed structures (e.g., PDB: 7ub5-a, 7ub6-c).
- The loss of the ΔV143, ΔY144, and ΔY145 mutations in BA2 introduces significant membrane binding propensity here in ACE-2-free structures where this element is resolved (e.g., PDB: 7ub5-a, 7ub5-ab, and 7ub6-a).
- The loss of the ΔN211, L212I, and ins214EPE mutations and the gain of the V213G mutation in the BA.2 NTD alters the flexibility and removes exposed hydrophobicity and charge in this mobile loop, which retains significant but variable and reduced membrane binding propensities in ACE-2-free structures.
- The T376A mutation in the BA.2 RTD decreased the membrane binding propensity of the neighbouring F375 residue from 67.2 ± 21.8 (PDB: 7tnw) to zero in all the BA.2 closed structures and induced conformational changes in this loop that reduces antibody interactions [7].
- The D405N and R408S mutations gained in BA.2 eliminate a negative and positive charge exposed in the RBM and balance the overall dipole moment. The former mutation is particularly critical for immune evasion [7], while the latter alters a binding pocket for fatty acid molecules of interest for its potential druggability [45,46,47].
- The loss of the G446S and G496S mutations found in BA.1 eliminates glycine residues that are common in membrane binding sites and allow flexibility during membrane insertion, although significant membrane binding propensities are retained around these positions.
- The D796Y mutation in BA.2 results in significant membrane binding propensity appearing in Y796 and I794 in approximately half of the structures.
- 7xix: all three V445 in chains A, B, and C,
- 7ub0: V16, N122, T124, F133, F140, L141 (chain B), and F486 (chain C),
- 7ub5: V483-F486 (chain A),
- 7ub6: F133, Q134, F140, L141 (chain B), V483, F486 (chain C), and
- 7xiw W152, F486 (chain D).
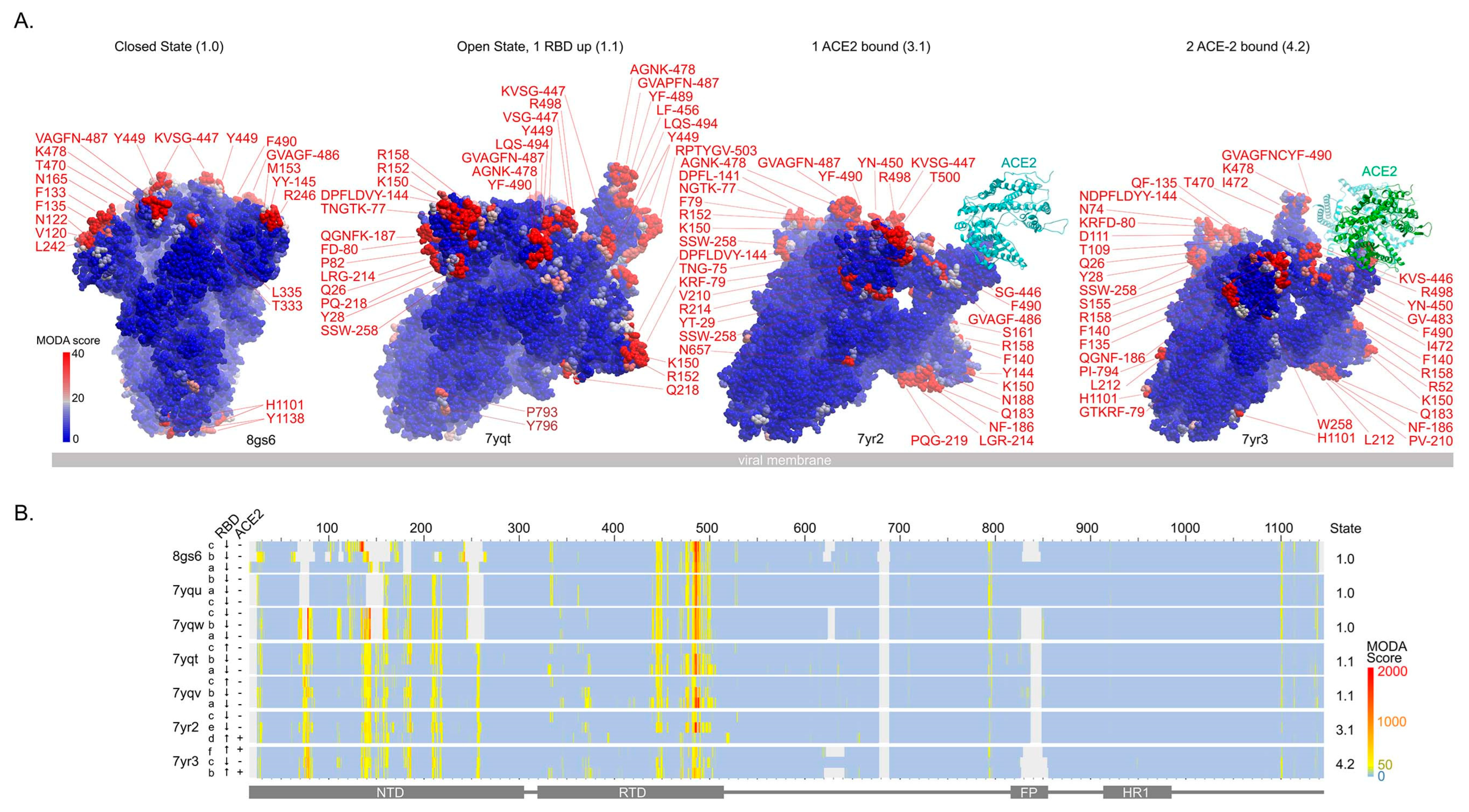
3.2. Conformations and Membrane Binding of BA.2.75 Spike Trimers
- The gain of the K147E mutation in BA.2.75 removes a positive charge from an exposed, dynamic loop, and membrane binding propensity position, which are present here, e.g., in BA.1 (7tof) and BA.2 (7xiw)
- The gain of the W152R mutation in BA.2.75 maintains membrane binding propensity at this NTD position in open and ACE-2-bound structures (PDBs: 7ypt, 7yqv, 7yr2, 3yr2), while in the closed structures, this element is disordered. However, the Trp at this position in BA.2 structures scores higher by MODA than the Arg here. The gain of a positive charge here compensates for the charge reversal at residue 147 that contributes to the dipole moment.
- The gain of the F157L mutation maintains membrane binding propensity at this NTD position in one subunit of BA.2.75 structures (PDBs: 7yqv, 7yr2). This residue is packed against Q14 and R158 (both of which are predicted to be membrane interactive by MODA) while being located at a C-terminus near a dynamic loop that is disordered in closed states. The Phe at this position scores higher by MODA in the BA.2 open structure (PDB: 7xiw), inferring greater membrane interactivity.
- The gain of the I210V mutation increases the membrane binding propensity at this NTD position in BA.2.75 structures (all but PDB 8gs6), with adjacent Pro209 and Leu212 often also being membrane interactive by MODA analysis.
- The gain of the G257S mutation in BA.2.75 maintains membrane binding propensity at this NTD position (as do neighbouring S256 and W258) in open and ACE-2-bound structures (PDBs: 7ypt, 7yqv, 7yr2, 3yr2), while in the closed structures this element is disordered.
- D339H is a novel mutation that induces local conformational changes, with this position packing against Phe371 [27]. This does not impart significant membrane binding propensity here in any BA.2.75 structure. The H339 sidechain is exposed at the top of the head, and is close to L335, which is membrane interactive (PDB: 8gs6, 7yqu, 7vqv, 7yr2). The positive charge at position 339 in low pH environments found along endocytic routes could favour membrane binding.
- The gain of the G446S mutation imparts resistance to antibodies [27] and maintains strong membrane binding propensity at this critical RBM position in all BA.2.75 trimers. This mutation compromises ACE-2 interactions [27], as well as flexibility that could be expected to facilitate membrane insertion.
- The gain of the N460K mutation in BA.2.75 contributes to the electrostatic dipole that favours membrane interactions but does not impart membrane binding propensity at this position in any BA.2.75 structures. Surprisingly, this mutation has been shown to either enhance ACE-2 affinity [27] or to have no such effect [28].
- R493Q maintains membrane binding propensity at this critical RBM position in a subset of BA.2.75 structures, and it also restores ACE-2 receptor affinity [6].
- 7yqt: F486 in chain A and S446, V483, G485, and F486 in chain B
- 7yqu: all three V445 in subunits A, B, and C,
- 7yqv: F486 in chain A; V483, G485, and F486 in chain B
- 7yqw: P793 and I794 in chain B
- 8gs6: V483, F486 in chain A, T345, K444, and V445 in B, and F133-F135, N165 in C.
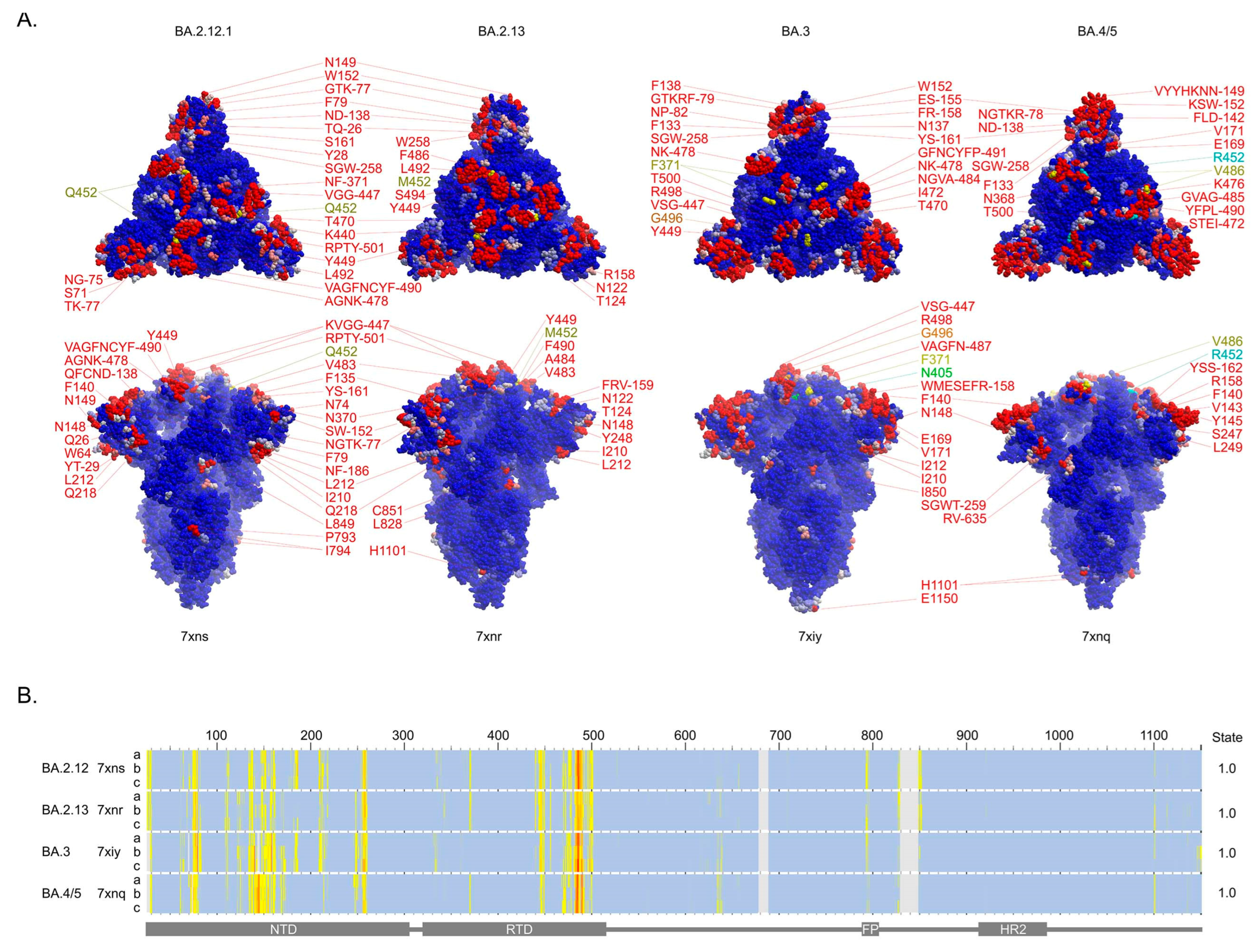
3.3. Membrane Binding by the Closed Spikes of BA.2.12, BA.2.13, BA.3 and BA.4/5 Variants
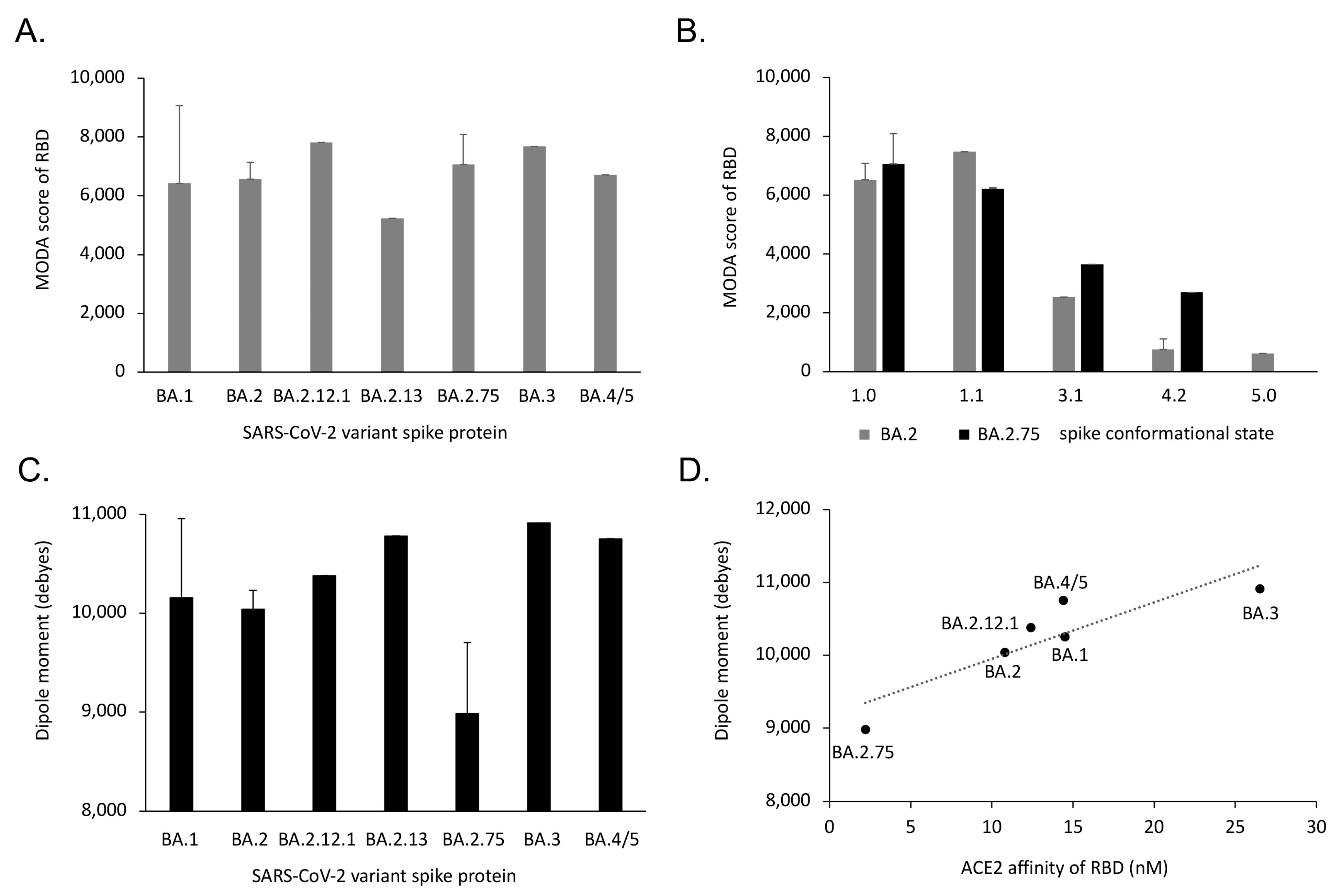
3.4. Comparison of Omicron Subvariant Interactions with Antibodies, Receptors and Membranes
3.5. Trends and Relationships of Evolutionary Drivers of Omicron Spikes
- Membrane docking propensity via the lipid binding surface of the spike head.
- ACE-2 receptor binding affinity via the RBM of the open state of the spike trimer.
- Neutralization by antibodies involved in immune escape.
- Electrostatic membrane attraction via the dipole moment of the spike trimer.
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Hui, K.P.Y.; Ho, J.C.W.; Cheung, M.C.; Ng, K.C.; Ching, R.H.H.; Lai, K.L.; Kam, T.T.; Gu, H.; Sit, K.Y.; Hsin, M.K.Y.; et al. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Satturwar, S.; Fowkes, M.; Farver, C.; Wilson, A.M.; Eccher, A.; Girolami, I.; Pujadas, E.; Bryce, C.; Salem, F.; El Jamal, S.M.; et al. Postmortem Findings Associated With SARS-CoV-2: Systematic Review and Meta-analysis. Am. J. Surg. Pathol. 2021, 45, 587–603. [Google Scholar] [CrossRef] [PubMed]
- Arora, P.; Nehlmeier, I.; Kempf, A.; Cossmann, A.; Schulz, S.R.; Dopfer-Jablonka, A.; Baier, E.; Tampe, B.; Moerer, O.; Dickel, S.; et al. Lung cell entry, cell-cell fusion capacity, and neutralisation sensitivity of omicron sublineage BA.2.75. Lancet Infect. Dis. 2022, 22, 1537–1538. [Google Scholar] [CrossRef]
- Iketani, S.; Liu, L.; Guo, Y.; Chan, J.F.; Huang, Y.; Wang, M.; Luo, Y.; Yu, J.; Chu, H.; Chik, K.K.; et al. Antibody evasion properties of SARS-CoV-2 Omicron sublineages. Nature 2022, 604, 553–556. [Google Scholar] [CrossRef]
- Wang, Q.; Guo, Y.; Iketani, S.; Nair, M.S.; Li, Z.; Mohri, H.; Wang, M.; Yu, J.; Bowen, A.D.; Chang, J.Y.; et al. Antibody evasion by SARS-CoV-2 Omicron subvariants BA.2.12.1, BA.4 and BA.5. Nature 2022, 608, 603–608. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X.; et al. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Zhang, N.; Wang, Y.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. BioRviv 2022, 1–3. (In Press) [Google Scholar] [CrossRef]
- Chen, C.; Nadeau, S.; Yared, M.; Voinov, P.; Xie, N.; Roemer, C.; Stadler, T. CoV-Spectrum: Analysis of Globally Shared SARS-CoV-2 Data to Identify and Characterize New Variants. Bioinformatics 2021, 38, 1735–1737. [Google Scholar] [CrossRef]
- Overduin, M.; Kervin, T.A.; Tran, A. Progressive membrane-binding mechanism of SARS-CoV-2 variant spike proteins. iScience 2022, 25, 104722. [Google Scholar] [CrossRef]
- Tran, A.; Kervin, T.A.; Overduin, M. Multifaceted membrane binding head of the SARS-CoV-2 spike protein. Curr. Res. Struct. Biol. 2022, 4, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, R.; Guest, J.D.; Yin, R.; Adolf-Bryfogle, J.; Schief, W.R.; Pierce, B.G. CoV3D: A database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021, 49, D282–D287. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein-ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef]
- Hong, Q.; Han, W.; Li, J.; Xu, S.; Wang, Y.; Xu, C.; Li, Z.; Zhang, C.; Huang, Z.; Cong, Y. Molecular basis of receptor binding and antibody neutralization of Omicron. Nature 2022, 604, 546–552. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Lavine, C.L.; Peng, H.; Zhu, H.; Anand, K.; Tong, P.; Gautam, A.; Mayer, M.L.; Rits-Volloch, S.; et al. Structural and functional impact by SARS-CoV-2 Omicron spike mutations. Cell. Rep. 2022, 39, 110729. [Google Scholar] [CrossRef] [PubMed]
- Gobeil, S.M.; Henderson, R.; Stalls, V.; Janowska, K.; Huang, X.; May, A.; Speakman, M.; Beaudoin, E.; Manne, K.; Li, D.; et al. Structural diversity of the SARS-CoV-2 Omicron spike. Mol. Cell 2022, 82, 2050–2068.e6. [Google Scholar] [CrossRef]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871.e813. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Zhou, J.; Tian, M.; Huang, M.; Liu, S.; Xie, Y.; Han, P.; Bai, C.; Zheng, A.; Fu, L.; et al. Omicron SARS-CoV-2 mutations stabilize spike up-RBD conformation and lead to a non-RBM-binding monoclonal antibody escape. Nat. Commun. 2022, 13, 4958. [Google Scholar] [CrossRef]
- Ye, G.; Liu, B.; Li, F. Cryo-EM structure of a SARS-CoV-2 omicron spike protein ectodomain. Nat. Commun. 2022, 13, 1214. [Google Scholar] [CrossRef]
- Ni, D.; Lau, K.; Turelli, P.; Raclot, C.; Beckert, B.; Nazarov, S.; Pojer, F.; Myasnikov, A.; Stahlberg, H.; Trono, D. Structural analysis of the Spike of the Omicron SARS-COV-2 variant by cryo-EM and implications for immune evasion. bioRxiv 2022. [Google Scholar] [CrossRef]
- Cerutti, G.; Guo, Y.; Liu, L.; Zhang, Z.; Luo, Y.; Huang, Y.; Wang, H.H.; Ho, D.D.; Sheng, Z.; Shapiro, L. Cryo-EM structure of the SARS-CoV-2 Omicron spike. Cell. Rep. 2022, 38, 110428. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, T.; Zhang, Y.; Yang, E.S.; Schramm, C.A.; Shi, W.; Pegu, A.; Oloninyi, O.K.; Ransier, A.; Darko, S.; et al. Antibodies with potent and broad neutralizing activity against antigenically diverse and highly transmissible SARS-CoV-2 variants. bioRxiv 2021. [Google Scholar] [CrossRef]
- Guo, H.; Gao, Y.; Li, T.; Lu, Y.; Zheng, L.; Liu, Y.; Yang, T.; Luo, F.; Song, S.; Wang, W.; et al. Structures of Omicron spike complexes and implications for neutralizing antibody development. Cell Rep. 2022, 39, 110770. [Google Scholar] [CrossRef]
- Xu, Y.; Wu, C.; Cao, X.; Gu, C.; Liu, H.; Jiang, M.; Wang, X.; Yuan, Q.; Wu, K.; Liu, J.; et al. Structural and biochemical mechanism for increased infectivity and immune evasion of Omicron BA.2 variant compared to BA.1 and their possible mouse origins. Cell Res. 2022, 32, 609–620. [Google Scholar] [CrossRef]
- Stalls, V.; Lindenberger, J.; Gobeil, S.M.; Henderson, R.; Parks, R.; Barr, M.; Deyton, M.; Martin, M.; Janowska, K.; Huang, X.; et al. Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike. Cell Rep. 2022, 39, 111009. [Google Scholar] [CrossRef]
- Saito, A.; Tamura, T.; Zahradnik, J.; Deguchi, S.; Tabata, K.; Anraku, Y.; Kimura, I.; Ito, J.; Yamasoba, D.; Nasser, H.; et al. Virological characteristics of the SARS-CoV-2 Omicron BA.2.75 variant. Cell Host Microbe 2022, 30, 1540–1555.e15. [Google Scholar] [CrossRef]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y.; et al. Characterization of the enhanced infectivity and antibody evasion of Omicron BA.2.75. Cell Host Microbe 2022, 30, 1527–1539.e5. [Google Scholar] [CrossRef]
- Chatzigoulas, A.; Cournia, Z. DREAMM: A web-based server for drugging protein-membrane interfaces as a novel workflow for targeted drug design. Bioinformatics 2022, 38, 5449–5451. [Google Scholar] [CrossRef]
- Chatzigoulas, A.; Cournia, Z. Predicting protein-membrane interfaces of peripheral membrane proteins using ensemble machine learning. Brief. Bioinform. 2022, 23, bbab518. [Google Scholar] [CrossRef]
- Schramm, C.A.; Hannigan, B.T.; Donald, J.E.; Keasar, C.; Saven, J.G.; Degrado, W.F.; Samish, I. Knowledge-based potential for positioning membrane-associated structures and assessing residue-specific energetic contributions. Structure 2012, 20, 924–935. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef]
- Lomize, A.L.; Todd, S.C.; Pogozheva, I.D. Spatial arrangement of proteins in planar and curved membranes by PPM 3.0. Protein. Sci. 2022, 31, 209–220. [Google Scholar] [CrossRef]
- Kufareva, I.; Lenoir, M.; Dancea, F.; Sridhar, P.; Raush, E.; Bissig, C.; Gruenberg, J.; Abagyan, R.; Overduin, M. Discovery of novel membrane binding structures and functions. Biochem. Cell Biol. 2014, 92, 555–563. [Google Scholar] [CrossRef]
- Bissig, C.; Lenoir, M.; Velluz, M.C.; Kufareva, I.; Abagyan, R.; Overduin, M.; Gruenberg, J. Viral infection controlled by a calcium-dependent lipid-binding module in ALIX. Dev. Cell 2013, 25, 364–373. [Google Scholar] [CrossRef]
- Bryant, J.A.; Morris, F.C.; Knowles, T.J.; Maderbocus, R.; Heinz, E.; Boelter, G.; Alodaini, D.; Colyer, A.; Wotherspoon, P.J.; Staunton, K.A.; et al. Structure of dual-BON domain protein DolP identifies phospholipid binding as a new mechanism for protein localization. Elife 2020, 9, e62614. [Google Scholar] [CrossRef]
- Overduin, M.; Kervin, T.A. The phosphoinositide code is read by a plethora of protein domains. Expert. Rev. Proteomics 2021, 18, 483–502. [Google Scholar] [CrossRef] [PubMed]
- Lenoir, M.; Kufareva, I.; Abagyan, R.; Overduin, M. Membrane and Protein Interactions of the Pleckstrin Homology Domain Superfamily. Membranes 2015, 5, 646–663. [Google Scholar] [CrossRef] [PubMed]
- Consortium, U. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef] [PubMed]
- Tuekprakhon, A.; Nutalai, R.; Dijokaite-Guraliuc, A.; Zhou, D.; Ginn, H.M.; Selvaraj, M.; Liu, C.; Mentzer, A.J.; Supasa, P.; Duyvesteyn, H.M.E.; et al. Antibody escape of SARS-CoV-2 Omicron BA.4 and BA.5 from vaccine and BA.1 serum. Cell 2022, 185, 2422–2433.e2413. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Raush, E.; Totrov, M.; Marsden, B.D.; Abagyan, R. A new method for publishing three-dimensional content. PLoS ONE 2009, 4, e7394. [Google Scholar] [CrossRef] [PubMed]
- Felder, C.E.; Prilusky, J.; Silman, I.; Sussman, J.L. A server and database for dipole moments of proteins. Nucleic Acids Res. 2007, 35, W512–W521. [Google Scholar] [CrossRef]
- Marcink, T.C.; Kicmal, T.; Armbruster, E.; Zhang, Z.; Zipursky, G.; Golub, K.L.; Idris, M.; Khao, J.; Drew-Bear, J.; McGill, G.; et al. Intermediates in SARS-CoV-2 spike-mediated cell entry. Sci. Adv. 2022, 8, eabo3153. [Google Scholar] [CrossRef] [PubMed]
- Toelzer, C.; Gupta, K.; Yadav, S.K.N.; Borucu, U.; Davidson, A.D.; Kavanagh Williamson, M.; Shoemark, D.K.; Garzoni, F.; Staufer, O.; Milligan, R.; et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science 2020, 370, 725–730. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Ye, F.; Guo, Y.; Xia, L.; Zhong, X.; Chi, X.; Zhou, Q. Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 2021, 31, 717–719. [Google Scholar] [CrossRef]
- Bangaru, S.; Ozorowski, G.; Turner, H.L.; Antanasijevic, A.; Huang, D.; Wang, X.; Torres, J.L.; Diedrich, J.K.; Tian, J.H.; Portnoff, A.D.; et al. Structural analysis of full-length SARS-CoV-2 spike protein from an advanced vaccine candidate. Science 2020, 370, 1089–1094. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; Dos Santos, M.S.; et al. SARS-CoV-2 can recruit a heme metabolite to evade antibody immunity. Sci. Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef]
- Yi, C.; Sun, X.; Lin, Y.; Gu, C.; Ding, L.; Lu, X.; Yang, Z.; Zhang, Y.; Ma, L.; Gu, W.; et al. Comprehensive mapping of binding hot spots of SARS-CoV-2 RBD-specific neutralizing antibodies for tracking immune escape variants. Genome Med. 2021, 13, 164. [Google Scholar] [CrossRef]
- Huo, J.; Dijokaite-Guraliuc, A.; Liu, C.; Zhou, D.; Ginn, H.M.; Das, R.; Supasa, P.; Selvaraj, M.; Nutalai, R.; Tuekprakhon, A.; et al. A delicate balance between antibody evasion and ACE2 affinity for Omicron BA.2.75. Cell Rep. 2022, 42, 111903. [Google Scholar] [CrossRef]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhong, C.; Tieleman, D.P. Supramolecular Organization of SARS-CoV and SARS-CoV-2 Virions Revealed by Coarse-Grained Models of Intact Virus Envelopes. J. Chem. Inf. Model 2021, 62, 176–186. [Google Scholar] [CrossRef]
- Wu, H.; Fujioka, Y.; Sakaguchi, S.; Suzuki, Y.; Nakano, T. Three-dimensional reconstruction by electron tomography for the application to ultrastructural analysis of SARS-CoV-2 particles. Med. Mol. Morphol. 2022, 55, 60–67. [Google Scholar] [CrossRef]
- Merolli, A.; Kasaei, L.; Ramasamy, S.; Kolloli, A.; Kumar, R.; Subbian, S.; Feldman, L.C. An intra-cytoplasmic route for SARS-CoV-2 transmission unveiled by Helium-ion microscopy. Sci. Rep. 2022, 12, 3794. [Google Scholar] [CrossRef]
- Asandei, A.; Mereuta, L.; Schiopu, I.; Park, J.; Seo, C.H.; Park, Y.; Luchian, T. Non-Receptor-Mediated Lipid Membrane Permeabilization by the SARS-CoV-2 Spike Protein S1 Subunit. ACS Appl. Mater. Interfaces 2020, 12, 55649–55658. [Google Scholar] [CrossRef]
- Luchini, A.; Micciulla, S.; Corucci, G.; Batchu, K.C.; Santamaria, A.; Laux, V.; Darwish, T.; Russell, R.A.; Thepaut, M.; Bally, I.; et al. Lipid bilayer degradation induced by SARS-CoV-2 spike protein as revealed by neutron reflectometry. Sci. Rep. 2021, 11, 14867. [Google Scholar] [CrossRef]
- Shoemark, D.K.; Colenso, C.K.; Toelzer, C.; Gupta, K.; Sessions, R.B.; Davidson, A.D.; Berger, I.; Schaffitzel, C.; Spencer, J.; Mulholland, A.J. Molecular Simulations suggest Vitamins, Retinoids and Steroids as Ligands of the Free Fatty Acid Pocket of the SARS-CoV-2 Spike Protein*. Angew Chem. Int. Ed. Engl. 2021, 60, 7098–7110. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Wan, L.; Yan, Q.; Wang, X.; Zhang, J.; Yang, X.; Zhang, Y.; Fan, C.; Li, D.; Deng, Y.; et al. HDL-scavenger receptor B type 1 facilitates SARS-CoV-2 entry. Nat. Metab. 2020, 2, 1391–1400. [Google Scholar] [CrossRef] [PubMed]
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.A.T.M.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef]
- Sanders, D.W.; Jumper, C.C.; Ackerman, P.J.; Bracha, D.; Donlic, A.; Kim, H.; Kenney, D.; Castello-Serrano, I.; Suzuki, S.; Tamura, T.; et al. SARS-CoV-2 requires cholesterol for viral entry and pathological syncytia formation. Elife 2021, 10, e65962. [Google Scholar] [CrossRef]
- Fantini, J.; Chahinian, H.; Yahi, N. Leveraging coronavirus binding to gangliosides for innovative vaccine and therapeutic strategies against COVID-19. Biochem. Biophys. Res. Commun. 2021, 538, 132–136. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Yahi, N.; Azzaz, F.; Chahinian, H. Structural dynamics of SARS-CoV-2 variants: A health monitoring strategy for anticipating Covid-19 outbreaks. J. Infect. 2021, 83, 197–206. [Google Scholar] [CrossRef]
- Raut, P.; Waters, H.; Zimmberberg, J.; Obeng, B.; Gosse, J.; Hess, S.T. Localization-Based Super-Resolution Microscopy Reveals Relationship between SARS-CoV2 Spike and Phosphatidylinositol (4,5)-bisphosphate. Proc. SPIE Int. Soc. Opt. Eng. 2022, 11965, 1196503. [Google Scholar] [PubMed]
- Carrique, L.; Duyvesteyn, H.M.; Malinauskas, T.; Zhao, Y.; Ren, J.; Zhou, D.; Walter, T.S.; Radecke, J.; Huo, J.; Ruza, R.R.; et al. The SARS-CoV-2 Spike Harbours a Lipid Binding Pocket Which Modulates Stability of the Prefusion Trimer. bioRxiv 2020. [Google Scholar] [CrossRef]
- Nguyen, L.; McCord, K.A.; Bui, D.T.; Bouwman, K.M.; Kitova, E.N.; Elaish, M.; Kumawat, D.; Daskhan, G.C.; Tomris, I.; Han, L.; et al. Sialic acid-containing glycolipids mediate binding and viral entry of SARS-CoV-2. Nat. Chem. Biol. 2021, 18, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.M.; Sandoval, D.R.; Spliid, C.B.; Pihl, J.; Perrett, H.R.; Painter, C.D.; Narayanan, A.; Majowicz, S.A.; Kwong, E.M.; McVicar, R.N.; et al. SARS-CoV-2 Infection Depends on Cellular Heparan Sulfate and ACE2. Cell 2020, 183, 1043–1057.e1015. [Google Scholar] [CrossRef]
- Shi, D.; Bu, C.; He, P.; Song, Y.; Dordick, J.S.; Linhardt, R.J.; Chi, L.; Zhang, F. Structural Characteristics of Heparin Binding to SARS-CoV-2 Spike Protein RBD of Omicron Sub-Lineages BA.2.12.1, BA.4 and BA.5. Viruses 2022, 14, 2696. [Google Scholar] [CrossRef]
- Chen, C.L.; Hasan, S.S.; Klose, T.; Sun, Y.; Buda, G.; Sun, C.; Klimstra, W.B.; Rossmann, M.G. Cryo-EM structure of eastern equine encephalitis virus in complex with heparan sulfate analogues. Proc. Natl. Acad. Sci. USA 2020, 117, 8890–8899. [Google Scholar] [CrossRef]
- Mendonça, L.; Howe, A.; Gilchrist, J.B.; Sheng, Y.; Sun, D.; Knight, M.L.; Zanetti-Domingues, L.C.; Bateman, B.; Krebs, A.S.; Chen, L.; et al. Correlative multi-scale cryo-imaging unveils SARS-CoV-2 assembly and egress. Nat. Commun. 2021, 12, 4629. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat. Commun. 2020, 11, 5885. [Google Scholar] [CrossRef]
- Caldas, L.A.; Carneiro, F.A.; Monteiro, F.L.; Augusto, I.; Higa, L.M.; Miranda, K.; Tanuri, A.; de Souza, W. Intracellular host cell membrane remodelling induced by SARS-CoV-2 infection in vitro. Biol. Cell. 2021, 113, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Eymieux, S.; Rouillé, Y.; Terrier, O.; Seron, K.; Blanchard, E.; Rosa-Calatrava, M.; Dubuisson, J.; Belouzard, S.; Roingeard, P. Ultrastructural modifications induced by SARS-CoV-2 in Vero cells: A kinetic analysis of viral factory formation, viral particle morphogenesis and virion release. Cell. Mol. Life Sci. 2021, 78, 3565–3576. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.L.; Rai, R.K.; Brown, J.C.; Griffin, P.; Edgar, J.R.; Shah, A.; Singanayagam, A.; Hogg, C.; Barclay, W.S.; Futter, C.E.; et al. Ultrastructural insight into SARS-CoV-2 entry and budding in human airway epithelium. Nat. Commun. 2022, 13, 1609. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | Residues That Are Mutated and Serve as Membrane Binders in Omicron Spike Trimer Structures | Next to a Membrane Binder in Sequence | Close in Space to a Membrane Binder |
|---|---|---|---|
| BA.1 | ΔH69, ΔV70, G142D, ΔV143, ΔY144, ΔY145, ΔN211, L212I, ins214EPE, G339D, S371L, S373P, S375F, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H | A67V, K417N | T95I |
| BA.2 | T19I, ΔL24, ΔP25, ΔP26, A27S, G142D, V213G, G339D, S371F, S373P, S375F, D405N, R408S, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H | T376A, K417N | |
| BA.2.12.1 | T19I, ΔL24, ΔP25, ΔP26, A27S, G142D, V213G, G339D, S371F, S373P, S375F, D405N, R408S, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H | T376A, K417N, L452Q | |
| BA.2.13 | T19I, ΔL24, ΔP25, ΔP26, A27S, G142D, V213G, G339D, S371F, S373P, S375F, D405N, R408S, N440K, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H | T376A, K417N, L452M | |
| BA.2.75 | T19I, ΔL24, ΔP25, ΔP26, A27S, G142D, K147E, W152R, F157L, I210V, V213G, G257S, G339H, S371F, S373P, S375F, D405N, R408S, N440K, G446S, N460K, S477N, T478K, E484A, R493Q, Q498R, N501Y, Y505H | T376A, K417N | |
| BA.3 | ΔH69, ΔV70, G142D, ΔV143, ΔY144, ΔY145, ΔN211, L212I, G339D, S371F, S373P, S375F, D405N, N440K, G446S, S477N, T478K, E484A, Q493R, Q498R, N501Y, Y505H | A67V, K417N | T95I |
| BA.4 | T19I, ΔL24, ΔP25, ΔP26, A27S, ΔH69, ΔV70, G142D, V213G, G339D, S371F, S373P, S375F, D405N, R408S, N440K, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H | T376A, K417N, L452R | |
| BA.5 | T19I, ΔL24, ΔP25, ΔP26, A27S, ΔH69, ΔV70, G142D, V213G, G339D, S371F, S373P, S375F, D405N, R408S, N440K, S477N, T478K, E484A, F486V, Q498R, N501Y, Y505H | T376A, K417N, L452R |
| Variant | RBDs | State | MODA | Dipole | Ligand | Resolution | PDB | Reference |
|---|---|---|---|---|---|---|---|---|
| BA.1 | 3 down | 1.0 | n.a. | 9758 | - | 2.56 | 7wp9 | [13] |
| BA.1 | down | 1.0 | n.a. | 7853 | - | 2.79 | 7t9j | [14] |
| BA.1 | 3 down | 1.0 | 4836 | 10765 | - | 3.10 | 7wk2 | [15] |
| BA.1 | 3 down | 1.0 | 3520 | 11247 | - | 3.10 | 7tnw | [16] |
| BA.1 | 3 down | 1.0 | 8660 | 9548 | - | 3.36 | 7tf8 | [17] |
| BA.1 | 3 down | 1.0 | 8676 | 9460 | - | 3.50 | 7tl1 | [17] |
| BA.1 | 3 down | 1.0 | 9198 | 11881 | - | 4.00 | 7wg7 | [18] |
| BA.1 | 1 up | 1.1 | 9510 | 11663 | - | 3.00 | 7y9s | [19] |
| BA.1 | 1 up | 1.1 | 9244 | 11403 | - | 3.00 | 7tgw | [20] |
| BA.1 | 1 up | 1.1 | 5382 | 12246 | - | 3.02 | 7qo7 | [21] |
| BA.1 | 1 up | 1.1 | 11654 | 6923 | - | 3.11 | 7thk | [22] |
| BA.1 | 1 up | 1.1 | 9475 | 10391 | - | 3.29 | 7tb4 | [23] |
| BA.1 | 1 up | 1.1 | 5329 | 11097 | - | 3.40 | 7wk3 | n.a. |
| BA.1 | 1 up | 1.1 | 9587 | 12937 | - | 3.40 | 7wg6 | [18] |
| BA.1 | 1 up | 1.1 | 6242 | 11702 | - | 3.40 | 7to4 | [16] |
| BA.1 | 1 up | 1.1 | n.a. | n.a. | - | 3.40 | 7tei | [17] |
| BA.1 | 1 up | 1.1 | 6237 | 11215 | - | 3.40 | 7wvn | [15] |
| BA.1 | 1 up | 1.1 | 3911 | 9486 | - | 3.41 | 7wvo | [15] |
| BA.1 | 1 up | 1.1 | 9654 | 10175 | - | 3.50 | 7tl9 | [17] |
| BA.1 | 1 up | 3.1 | 1885 | 4158 | 1 ACE-2 | 2.77 | 7wpa | [13] |
| BA.1 | 1 up | 3.1 | 4368 | 4393 | 1 ACE-2 | 2.85 | 7y9z | [19] |
| BA.1 | 1 up | 3.1 | 2440 | 2511 | 1 ACE-2 | 2.90 | 7ws9 | [24] |
| BA.1 | 1 up | 3.1 | 3085 | 3906 | 1 ACE-2 | 3.13 | 7xo5 | [25] |
| BA.1 | 1 up | 3.1 | 3423 | 4672 | 1 ACE-2 | 3.69 | 7wk4 | [15] |
| BA.1 | 2 up | 3.2 | 5248 | 5256 | 1 ACE-2 | 3.66 | 7wk5 | [15] |
| BA.1 | 2 up | 3.2 | 5261 | 5243 | 1 ACE-2 | 3.70 | 7wvp | [15] |
| BA.1 | 3 up | 3.3 | 7420 | 5770 | 1 ACE-2 | 4.04 | 7wvq | [15] |
| BA.1 | 2 up | 4.2 | 542 | n.a. | 2 ACE-2 | 2.45 | 7t9k | [14] |
| BA.1 | 2 up | 4.2 | 1070 | 7679 | 2 ACE-2 | 3.00 | 7ws8 | [24] |
| BA.1 | 2 up | 4.2 | 953 | 4278 | 2 ACE-2 | 3.24 | 7xo4 | [25] |
| BA.1 | 2 up | 4.2 | 2523 | 9770 | 2 ACE-2 | 3.30 | 7xid | [25] |
| BA.1 | 2 up | 4.2 | 2448 | 8324 | 2 ACE-2 | 3.40 | 7xch | [19] |
| BA.1 | 2 up | 4.2 | 1663 | 8544 | 2 ACE-2 | 3.50 | 7wgb | [18] |
| BA.1 | 3 up | 5.0 | 112 | 12925 | 3 ACE-2 | 3.10 | 7ya0 | [19] |
| BA.2 | 3 down | 1.0 | 7252 | 10081 | - | 3.25 | 7xix | [7] |
| BA.2 | 3 down | 1.0 | 6356 | 10173 | - | 3.31 | 7ub0 | [26] |
| BA.2 | 3 down | 1.0 | 5908 | 9760 | - | 3.35 | 7ub5 | [26] |
| BA.2 | 3 down | 1.0 | 6739 | 10146 | - | 3.52 | 7ub6 | [26] |
| BA.2 | 1 up | 1.1 | 7481 | 11259 | - | 3.62 | 7xiw | [7] |
| BA.2 | 1 up | 3.1 | 2537 | 3265 | 1 ACE-2 | 3.20 | 7xoa | [25] |
| BA.2 | 2 up | 4.2 | 511 | 4683 | 2 ACE-2 | 3.30 | 7xob | [25] |
| BA.2 | 2 up | 4.2 | 1011 | 7916 | 2 ACE-2 | 3.38 | 7xo7 | [25] |
| BA.2 | 3 up | 5.0 | 619 | 12139 | 3 ACE-2 | 3.48 | 7xo8 | [25] |
| BA.2.12.1 | 3 down | 1.0 | 7811 | 10381 | - | 3.48 | 7xns | [7] |
| BA.2.13 | 3 down | 1.0 | 5230 | 10781 | - | 3.49 | 7xnr | [7] |
| BA.2.75 | 3 down | 1.0 | 8214 | 8158 | - | 2.86 | 8gs6 | [27] |
| BA.2.75 | 3 down | 1.0 | 6241 | 9500 | - | 3.19 | 7yqu | [28] |
| BA.2.75 | 3 down | 1.0 | 6732 | 9285 | - | 3.51 | 7yqw | [28] |
| BA.2.75 | 1 up | 1.1 | 6246 | 9661 | - | 3.45 | 7yqt | [28] |
| BA.2.75 | 1 up | 1.1 | 6202 | 9646 | - | 3.58 | 7yqv | [28] |
| BA.2.75 | 1 up | 3.1 | 3649 | 6056 | 1 ACE-2 | 3.30 | 7yr2 | [28] |
| BA.2.75 | 2 up | 4.2 | 2693 | 10929 | 2 ACE-2 | 3.52 | 7yr3 | [28] |
| BA.3 | 3 down | 1.0 | 7674 | 10912 | - | 3.07 | 7xiy | [7] |
| BA.4/5 | 3 down | 1.0 | 6715 | 10753 | - | 3.52 | 7xnq | [7] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Overduin, M.; Bhat, R.K.; Kervin, T.A. SARS-CoV-2 Omicron Subvariants Balance Host Cell Membrane, Receptor, and Antibody Docking via an Overlapping Target Site. Viruses 2023, 15, 447. https://doi.org/10.3390/v15020447
Overduin M, Bhat RK, Kervin TA. SARS-CoV-2 Omicron Subvariants Balance Host Cell Membrane, Receptor, and Antibody Docking via an Overlapping Target Site. Viruses. 2023; 15(2):447. https://doi.org/10.3390/v15020447
Chicago/Turabian StyleOverduin, Michael, Rakesh K. Bhat, and Troy A. Kervin. 2023. "SARS-CoV-2 Omicron Subvariants Balance Host Cell Membrane, Receptor, and Antibody Docking via an Overlapping Target Site" Viruses 15, no. 2: 447. https://doi.org/10.3390/v15020447