
When Plaquing Is Not Possible: Computational Methods for Detecting Induced Phages

Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Genome Sequencing, and Genome Assembly
2.2. Prophage Sequence Prediction
2.3. Culturing Bacteria for Spontaneous Induction and Sequencing
2.4. Mock Community Construction
2.5. Identification of Positive Control Induced Phage
2.6. Identification of Induced Phage in a Metagenome Data Set
2.7. Prophage Induction Estimator (PIE)
3. Results
3.1. Computational Method for Detecting Induced Prophages
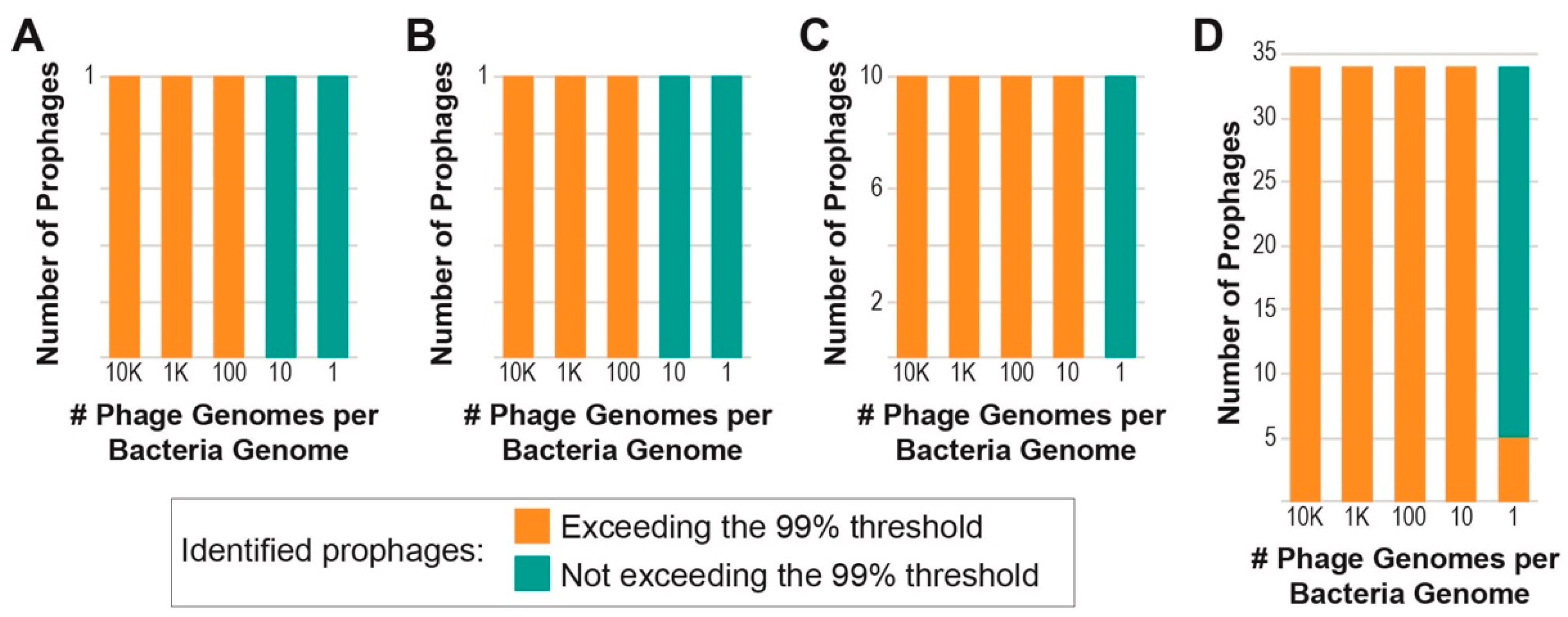
3.2. Assessing PIE’s Ability to Detect Induced Prophages on In Silico Data
3.3. Proof-of-Concept: Detecting Induced Phages
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cobián Güemes, A.G.; Youle, M.; Cantú, V.A.; Felts, B.; Nulton, J.; Rohwer, F. Viruses as Winners in the Game of Life. Annu. Rev. Virol. 2016, 3, 197–214. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Bushman, F.D. The Human Virome: Assembly, Composition and Host Interactions. Nat. Rev. Microbiol. 2021, 19, 514–527. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going Viral: Next-Generation Sequencing Applied to Phage Populations in the Human Gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Aggarwala, V.; Liang, G.; Bushman, F.D. Viral Communities of the Human Gut: Metagenomic Analysis of Composition and Dynamics. Mob. DNA 2017, 8, 12. [Google Scholar] [CrossRef] [PubMed]
- Barr, J.J. A Bacteriophages Journey through the Human Body. Immunol. Rev. 2017, 279, 106–122. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Hill, C. Bacteriophages of the Human Gut: The “Known Unknown” of the Microbiome. Cell Host Microbe 2019, 25, 195–209. [Google Scholar] [CrossRef]
- Szafrański, S.P.; Slots, J.; Stiesch, M. The Human Oral Phageome. Periodontol. 2000 2021, 86, 79–96. [Google Scholar] [CrossRef]
- Garretto, A.; Miller-Ensminger, T.; Wolfe, A.J.; Putonti, C. Bacteriophages of the Lower Urinary Tract. Nat. Rev. Urol. 2019, 16, 422–432. [Google Scholar] [CrossRef]
- Boxberger, M.; Cenizo, V.; Cassir, N.; La Scola, B. Challenges in Exploring and Manipulating the Human Skin Microbiome. Microbiome 2021, 9, 125. [Google Scholar] [CrossRef]
- Hatfull, G.F.; Hendrix, R.W. Bacteriophages and Their Genomes. Curr. Opin. Virol. 2011, 1, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic Analyses of an Uncultured Viral Community from Human Feces. J. Bacteriol. 2003, 185, 6220–6223. [Google Scholar] [CrossRef]
- Reyes, A.; Haynes, M.; Hanson, N.; Angly, F.E.; Heath, A.C.; Rohwer, F.; Gordon, J.I. Viruses in the Faecal Microbiota of Monozygotic Twins and Their Mothers. Nature 2010, 466, 334–338. [Google Scholar] [CrossRef]
- Minot, S.; Sinha, R.; Chen, J.; Li, H.; Keilbaugh, S.A.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. The Human Gut Virome: Inter-Individual Variation and Dynamic Response to Diet. Genome Res. 2011, 21, 1616–1625. [Google Scholar] [CrossRef]
- Pride, D.T.; Salzman, J.; Haynes, M.; Rohwer, F.; Davis-Long, C.; White, R.A.; Loomer, P.; Armitage, G.C.; Relman, D.A. Evidence of a Robust Resident Bacteriophage Population Revealed through Analysis of the Human Salivary Virome. ISME J. 2012, 6, 915–926. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Meisel, J.S.; Tyldsley, A.S.; Zheng, Q.; Hodkinson, B.P.; SanMiguel, A.J.; Minot, S.; Bushman, F.D.; Grice, E.A. The Human Skin Double-Stranded DNA Virome: Topographical and Temporal Diversity, Genetic Enrichment, and Dynamic Associations with the Host Microbiome. MBio 2015, 6, e01578-15. [Google Scholar] [CrossRef]
- Miller-Ensminger, T.; Garretto, A.; Brenner, J.; Thomas-White, K.; Zambom, A.; Wolfe, A.J.; Putonti, C. Bacteriophages of the Urinary Microbiome. J. Bacteriol. 2018, 200, e00738-17. [Google Scholar] [CrossRef]
- Nanda, A.M.; Thormann, K.; Frunzke, J. Impact of Spontaneous Prophage Induction on the Fitness of Bacterial Populations and Host-Microbe Interactions. J. Bacteriol. 2015, 197, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.P.; Jacob, F.; Goldthwait, D.A. Prophage Induction and Filament Formation in a Mutant Strain of Escherichia Coli. Proc. Natl. Acad. Sci. USA 1967, 58, 1903–1910. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, B.J.; Cox, S.H.; Jett, J.H. Prophage Induction and Inactivation by UV Light. J. Virol. 1976, 18, 950–955. [Google Scholar] [CrossRef] [PubMed]
- Meijer, W.; van de Bunt, B.; Twigt, M.; de Jonge, B.; Smit, G.; Hugenholtz, J. Lysis of Lactococcus Lactis Subsp. Cremoris SK110 and Its Nisin-Immune Transconjugant in Relation to Flavor Development in Cheese. Appl. Environ. Microbiol. 1998, 64, 1950–1953. [Google Scholar] [CrossRef] [Green Version]
- Lunde, M.; Aastveit, A.H.; Blatny, J.M.; Nes, I.F. Effects of Diverse Environmental Conditions on ΦLC3 Prophage Stability in Lactococcus Lactis. Appl. Environ. Microbiol. 2005, 71, 721–727. [Google Scholar] [CrossRef]
- Williamson, S.J.; Paul, J.H. Environmental Factors That Influence the Transition from Lysogenic to Lytic Existence in the PhiHSIC/Listonella Pelagia Marine Phage-Host System. Microb. Ecol. 2006, 52, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Wallin-Carlquist, N.; Cao, R.; Márta, D.; da Silva, A.S.; Schelin, J.; Rådström, P. Acetic Acid Increases the Phage-Encoded Enterotoxin A Expression in Staphylococcus Aureus. BMC Microbiol. 2010, 10, 147. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kotay, S.M.; Goel, R. Various Physico-Chemical Stress Factors Cause Prophage Induction in Nitrosospira Multiformis 25196—An Ammonia Oxidizing Bacteria. Water Res. 2010, 44, 4550–4558. [Google Scholar] [CrossRef]
- Chu, T.-C.; Murray, S.R.; Hsu, S.-F.; Vega, Q.; Lee, L.H. Temperature-Induced Activation of Freshwater Cyanophage AS-1 Prophage. Acta Histochem. 2011, 113, 294–299. [Google Scholar] [CrossRef]
- Zeaki, N.; Rådström, P.; Schelin, J. Evaluation of Potential Effects of NaCl and Sorbic Acid on Staphylococcal Enterotoxin A Formation. Microorganisms 2015, 3, 551–566. [Google Scholar] [CrossRef]
- Miller-Ensminger, T.; Garretto, A.; Stark, N.; Putonti, C. Mimicking Prophage Induction in the Body: Induction in the Lab with PH Gradients. PeerJ 2020, 8, e9718. [Google Scholar] [CrossRef]
- Silveira, C.B.; Luque, A.; Rohwer, F. The Landscape of Lysogeny across Microbial Community Density, Diversity and Energetics. Environ. Microbiol. 2021, 23, 4098–4111. [Google Scholar] [CrossRef]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e5. [Google Scholar] [CrossRef]
- Koskella, B.; Meaden, S. Understanding Bacteriophage Specificity in Natural Microbial Communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Viral Community Function from Genomic Sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef] [PubMed]
- Tisza, M.J.; Belford, A.K.; Domínguez-Huerta, G.; Bolduc, B.; Buck, C.B. Cenote-Taker 2 Democratizes Virus Discovery and Sequence Annotation. Virus Evol. 2021, 7, veaa100. [Google Scholar] [CrossRef] [PubMed]
- Waller, A.S.; Yamada, T.; Kristensen, D.M.; Kultima, J.R.; Sunagawa, S.; Koonin, E.V.; Bork, P. Classification and Quantification of Bacteriophage Taxa in Human Gut Metagenomes. ISME J. 2014, 8, 1391–1402. [Google Scholar] [CrossRef]
- Delbrück, M. The Burst Size Distribution in the Growth of Bacterial Viruses (Bacteriophages). J. Bacteriol. 1945, 50, 131–135. [Google Scholar] [CrossRef]
- Warner, H.R.; Drong, R.F.; Berget, S.M. Early Events after Infection of Escherichia Coli by Bacteriophage T5. Induction of a 5′-Nucleotidase Activity and Excretion of Free Bases. J. Virol. 1975, 15, 273–280. [Google Scholar] [CrossRef]
- Turkington, C.J.R.; Abadi, N.N.; Edwards, R.A.; Grasis, J.A. hafeZ: Active Prophage Identification through Read Mapping. bioRxiv 2021, 2021-07. [Google Scholar] [CrossRef]
- Kieft, K.; Anantharaman, K. Deciphering Active Prophages from Metagenomes. mSystems 2022, 7, e0008422. [Google Scholar] [CrossRef]
- Zünd, M.; Ruscheweyh, H.-J.; Field, C.M.; Meyer, N.; Cuenca, M.; Hoces, D.; Hardt, W.-D.; Sunagawa, S. High Throughput Sequencing Provides Exact Genomic Locations of Inducible Prophages and Accurate Phage-to-Host Ratios in Gut Microbial Strains. Microbiome 2021, 9, 77. [Google Scholar] [CrossRef]
- Price, T.K.; Dune, T.; Hilt, E.E.; Thomas-White, K.J.; Kliethermes, S.; Brincat, C.; Brubaker, L.; Wolfe, A.J.; Mueller, E.R.; Schreckenberger, P.C. The Clinical Urine Culture: Enhanced Techniques Improve Detection of Clinically Relevant Microorganisms. J. Clin. Microbiol. 2016, 54, 1216–1222. [Google Scholar] [CrossRef]
- Garretto, A.; Miller-Ensminger, T.; Ene, A.; Merchant, Z.; Shah, A.; Gerodias, A.; Biancofiori, A.; Canchola, S.; Canchola, S.; Castillo, E.; et al. Genomic Survey of E. Coli From the Bladders of Women with and without Lower Urinary Tract Symptoms. Front. Microbiol. 2020, 11, 2094. [Google Scholar] [CrossRef] [PubMed]
- Markovic, T.; Miller-Ensminger, T.; Voukadinova, A.; Wolfe, A.J.; Putonti, C. Draft Genome Sequence of Actinomyces neuii UMB1295, Isolated from the Female Urinary Tract. Microbiol. Resour. Announc. 2020, 9, e00402-20. [Google Scholar] [CrossRef] [PubMed]
- Putonti, C.; Ahmad, A.; Baddoo, G.; Diaz, J.; Do, M.; Gallian, N.; Lorentzen, C.; Mohammed, H.; Murphy, J.; Olu-Ajeigbe, A.; et al. Draft Genome Sequences of 11 Lactobacillus Jensenii Strains Isolated from the Female Bladder. Microbiol. Resour. Announc. 2019, 8, e00970-19. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.; Miller-Ensminger, T.; Voukadinova, A.; Wolfe, A.J.; Putonti, C. Draft Genome Sequence of Enterococcus Faecalis UMB1309, Isolated from Catheterized Urine. Microbiol. Resour. Announc. 2020, 9, e00406-20. [Google Scholar] [CrossRef]
- Allen, O.; Miller-Ensminger, T.; Voukadinova, A.; Wolfe, A.J.; Putonti, C. Draft Genome Sequence of Proteus Mirabilis UMB1310, Isolated from the Female Urinary Tract. Microbiol. Resour. Announc. 2020, 9, e00390-20. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-Generation Sequencing Read Simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Johnson, G.; Stark, N.; Wolfe, A.J.; Putonti, C. Complete Genome Sequence of a Pseudomonas Aeruginosa Isolate from a Kidney Stone. Microbiol. Resour. Announc. 2019, 8, e01073-19. [Google Scholar] [CrossRef]
- Johnson, G.; Wolfe, A.J.; Putonti, C. Characterization of the ΦCTX-like Pseudomonas Aeruginosa Phage Dobby Isolated from the Kidney Stone Microbiota. Access Microbiol. 2019, 1, e000002. [Google Scholar] [CrossRef]
- Mormando, R. Polyomavirus JCPyV and Its Role in the Urinary Tract; Loyola University Chicago: Chicago, IL, USA, 2022. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A New Versatile Metagenomic Assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A Resource Combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef] [PubMed]
- Silverman, B.W. Density Estimation for Statistics and Data Analysis; Monographs on statistics and applied probability; Chapman & Hall/CRC: Boca Raton, FL, USA, 1998; ISBN 978-0-412-24620-3. [Google Scholar]
- Tisza, M.J.; Buck, C.B. A Catalog of Tens of Thousands of Viruses from Human Metagenomes Reveals Hidden Associations with Chronic Diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2023202118. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, S.I.; Kiliç, A.O.; Mou, S.M.; Tao, L. Phage Infection in Vaginal Lactobacilli: An in Vitro Study. Infect. Dis. Obstet. Gynecol. 1997, 5, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-H.; Alexander, L.M.; Pan, M.; Schueler, K.L.; Keller, M.P.; Attie, A.D.; Walter, J.; van Pijkeren, J.-P. Dietary Fructose and Microbiota-Derived Short-Chain Fatty Acids Promote Bacteriophage Production in the Gut Symbiont Lactobacillus Reuteri. Cell Host Microbe 2019, 25, 273–284.e6. [Google Scholar] [CrossRef]
- Paez-Espino, D.; Eloe-Fadrosh, E.A.; Pavlopoulos, G.A.; Thomas, A.D.; Huntemann, M.; Mikhailova, N.; Rubin, E.; Ivanova, N.N.; Kyrpides, N.C. Uncovering Earth’s Virome. Nature 2016, 536, 425–430. [Google Scholar] [CrossRef]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Matheus Carnevali, P.B.; Cheng, J.-F.; Ivanova, N.N.; et al. Cryptic Inoviruses Revealed as Pervasive in Bacteria and Archaea across Earth’s Biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef]
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of Huge Phages from across Earth’s Ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- Jian, H.; Yi, Y.; Wang, J.; Hao, Y.; Zhang, M.; Wang, S.; Meng, C.; Zhang, Y.; Jing, H.; Wang, Y.; et al. Diversity and Distribution of Viruses Inhabiting the Deepest Ocean on Earth. ISME J. 2021, 15, 3094–3110. [Google Scholar] [CrossRef]
- Camarillo-Guerrero, L.F.; Almeida, A.; Rangel-Pineros, G.; Finn, R.D.; Lawley, T.D. Massive Expansion of Human Gut Bacteriophage Diversity. Cell 2021, 184, 1098–1109.e9. [Google Scholar] [CrossRef] [PubMed]
- Zayed, A.A.; Wainaina, J.M.; Dominguez-Huerta, G.; Pelletier, E.; Guo, J.; Mohssen, M.; Tian, F.; Pratama, A.A.; Bolduc, B.; Zablocki, O.; et al. Cryptic and Abundant Marine Viruses at the Evolutionary Origins of Earth’s RNA Virome. Science 2022, 376, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.G.; Russ, C.; Costello, M.; Hollinger, A.; Lennon, N.J.; Hegarty, R.; Nusbaum, C.; Jaffe, D.B. Characterizing and Measuring Bias in Sequence Data. Genome Biol. 2013, 14, R51. [Google Scholar] [CrossRef]
- Grazziotin, A.L.; Koonin, E.V.; Kristensen, D.M. Prokaryotic Virus Orthologous Groups (PVOGs): A Resource for Comparative Genomics and Protein Family Annotation. Nucleic Acids Res. 2017, 45, D491–D498. [Google Scholar] [CrossRef] [PubMed]
- Paterson, S.; Vogwill, T.; Buckling, A.; Benmayor, R.; Spiers, A.J.; Thomson, N.R.; Quail, M.; Smith, F.; Walker, D.; Libberton, B.; et al. Antagonistic Coevolution Accelerates Molecular Evolution. Nature 2010, 464, 275–278. [Google Scholar] [CrossRef]
- Chevallereau, A.; Pons, B.J.; van Houte, S.; Westra, E.R. Interactions between Bacterial and Phage Communities in Natural Environments. Nat. Rev. Microbiol. 2022, 20, 49–62. [Google Scholar] [CrossRef]
- Fortier, L.-C.; Sekulovic, O. Importance of Prophages to Evolution and Virulence of Bacterial Pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef]
- Obeng, N.; Pratama, A.A.; Elsas, J.D. van The Significance of Mutualistic Phages for Bacterial Ecology and Evolution. Trends Microbiol. 2016, 24, 440–449. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | SRA Accession No. | GenBank Assembly Accession No. | Culture Media |
|---|---|---|---|
| E. coli UMB1284 | SRR7534266 | GCA_003892355.1 | LB |
| A. neuii UMB1295 | SRR11441016 | GCA_012030015.1 | Actinomyces Broth |
| S. hominus UMB1296-1T | SRR14752304 | GCA_018919365.1 | TSB |
| L. jensenii UMB1303 | SRR9695709 | GCA_007786145.1 | MRS + 5% Tween80 |
| E. faecalis UMB1309 | SRR11441014 | GCA_012030535.1 | BHI |
| P. mirabilis UMB1310 | SRR11441013 | GCA_012030515.1 | LB |
| C. amycolatum UMB1310-1E | SRR14752303 | GCA_018919345.1 | LB |
| Contig ID | Length | Coverage | BLAST Best Hit | |||
|---|---|---|---|---|---|---|
| Description | Query Cov | %ID | Accession No. | |||
| NODE_65 | 9541 | 536.29 | JC polyomavirus | 100 | 97.33 | LT615220.1 |
| NODE_24 | 37847 | 215.32 | TPA: Siphoviridae sp. ctX581 | 100 | 99.36 | BK014883.1 |
| NODE_1047 | 1563 | 115.85 | L. gasseri plasmid pHL20_1 | 92 | 99.72 | CP072179.1 |
| NODE_15 | 61268 | 109.23 | Lactobacillus iners KY 1 | 86 | 99.06 | CP048798.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miller-Ensminger, T.; Johnson, G.; Banerjee, S.; Putonti, C. When Plaquing Is Not Possible: Computational Methods for Detecting Induced Phages. Viruses 2023, 15, 420. https://doi.org/10.3390/v15020420
Miller-Ensminger T, Johnson G, Banerjee S, Putonti C. When Plaquing Is Not Possible: Computational Methods for Detecting Induced Phages. Viruses. 2023; 15(2):420. https://doi.org/10.3390/v15020420
Chicago/Turabian StyleMiller-Ensminger, Taylor, Genevieve Johnson, Swarnali Banerjee, and Catherine Putonti. 2023. "When Plaquing Is Not Possible: Computational Methods for Detecting Induced Phages" Viruses 15, no. 2: 420. https://doi.org/10.3390/v15020420