
Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID


Abstract
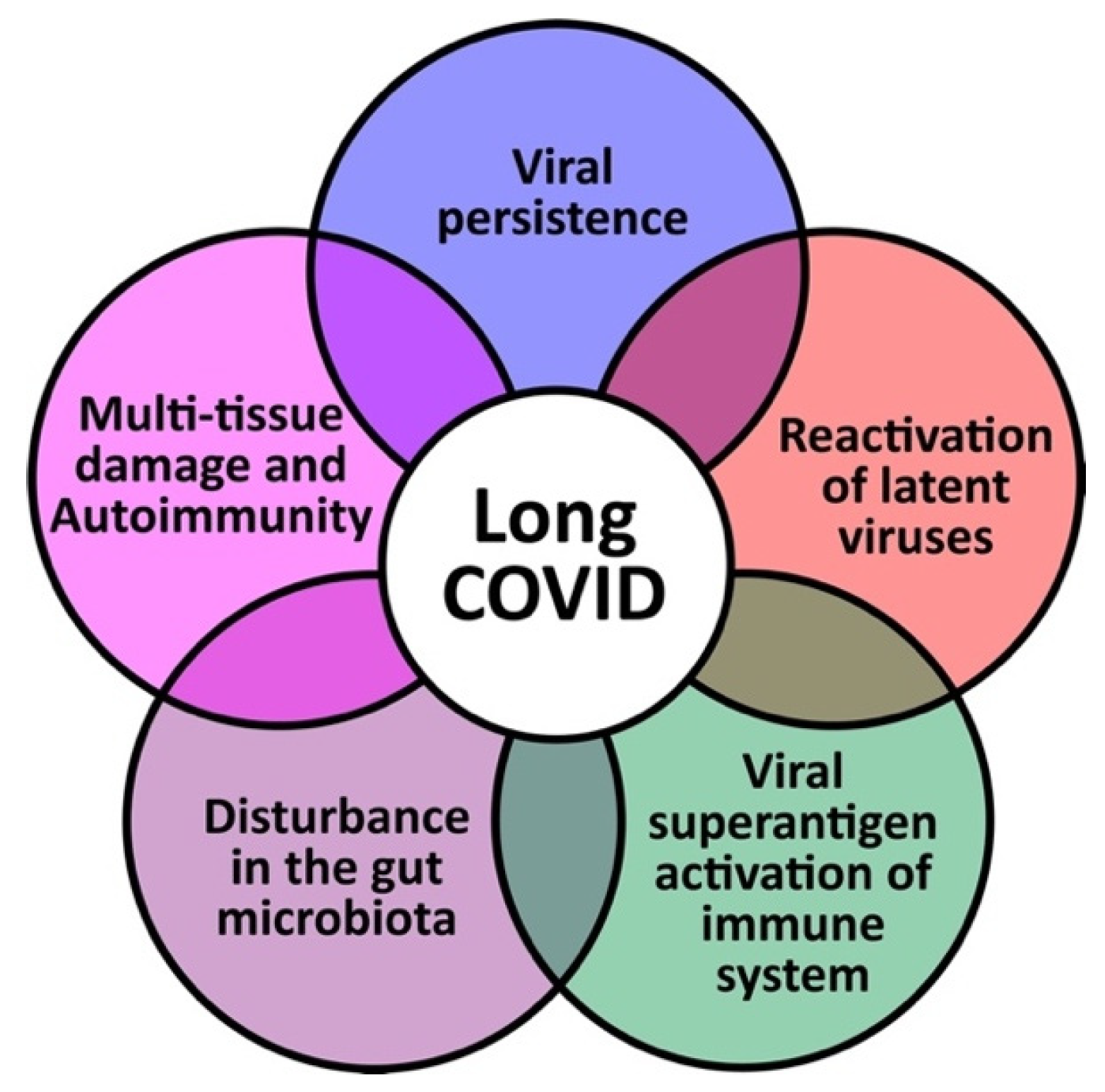
:1. Introduction
2. Viral Persistence
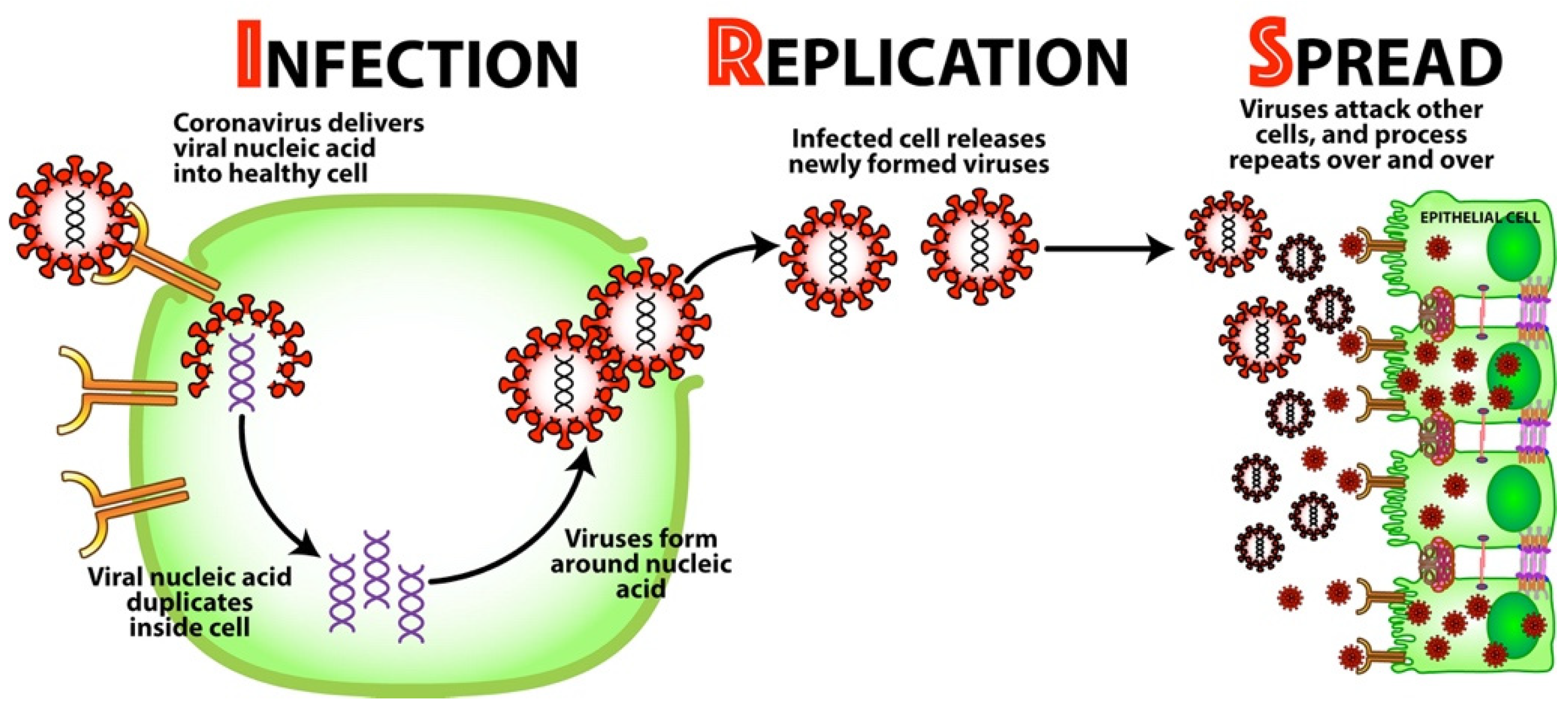
2.1. Background: Mechanism for Viral Infection
- Infection—Once bound to the receptor, the virus delivers its viral nucleic acid into the healthy cell.
- Replication—The viral nucleic acid duplicates inside the cell, and the cell’s resources are used to form more viruses around the nucleic acid. The now-infected cell releases these newly formed viruses.
- Spread—The newly formed viruses now go on to attack other cells all throughout the body, and the process repeats over and over.
2.2. What Is Viral Persistence?
3. Reactivation of Latent Viruses
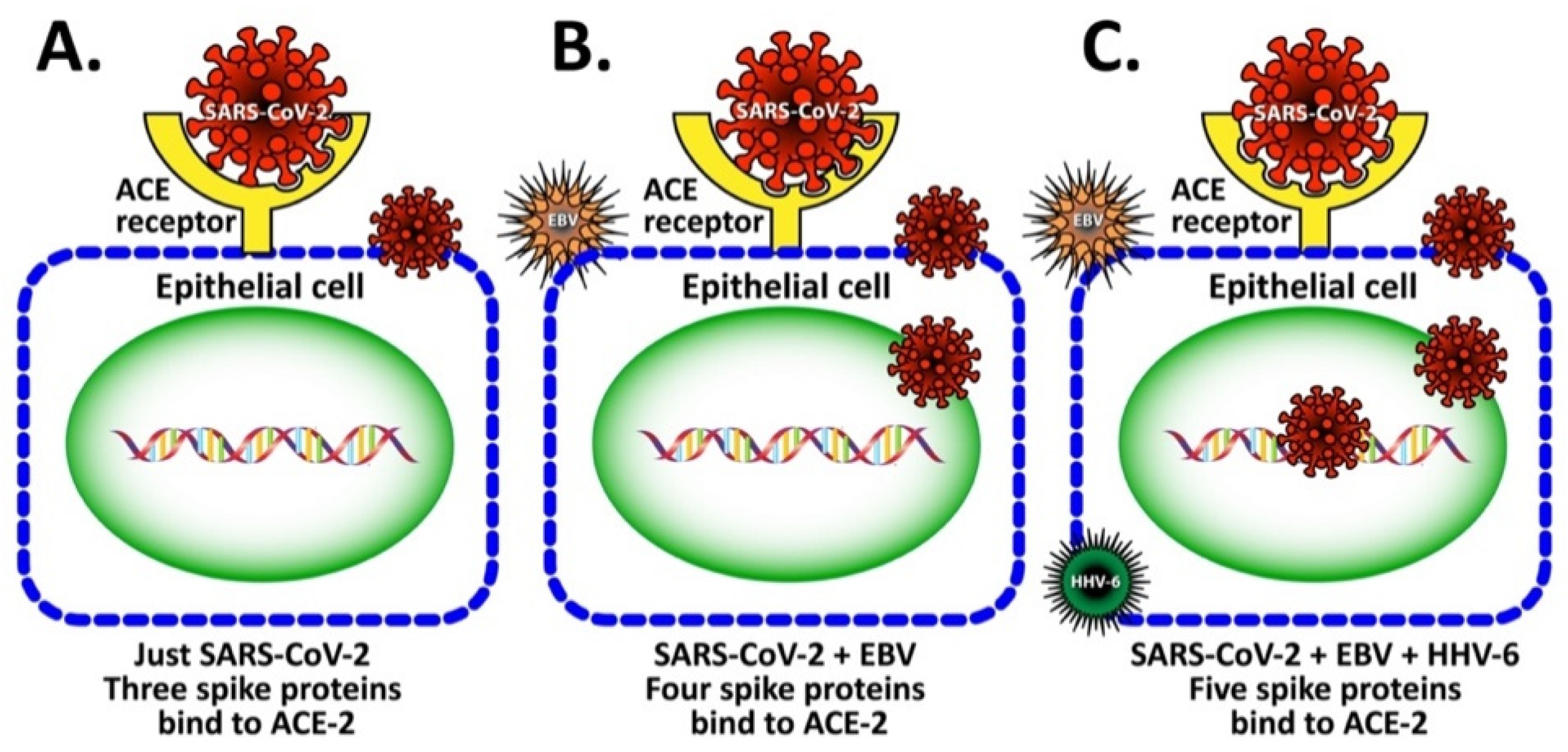
3.1. Mechanism by Which EBV Participates in the Enhancement of SARS-CoV-2 Infection
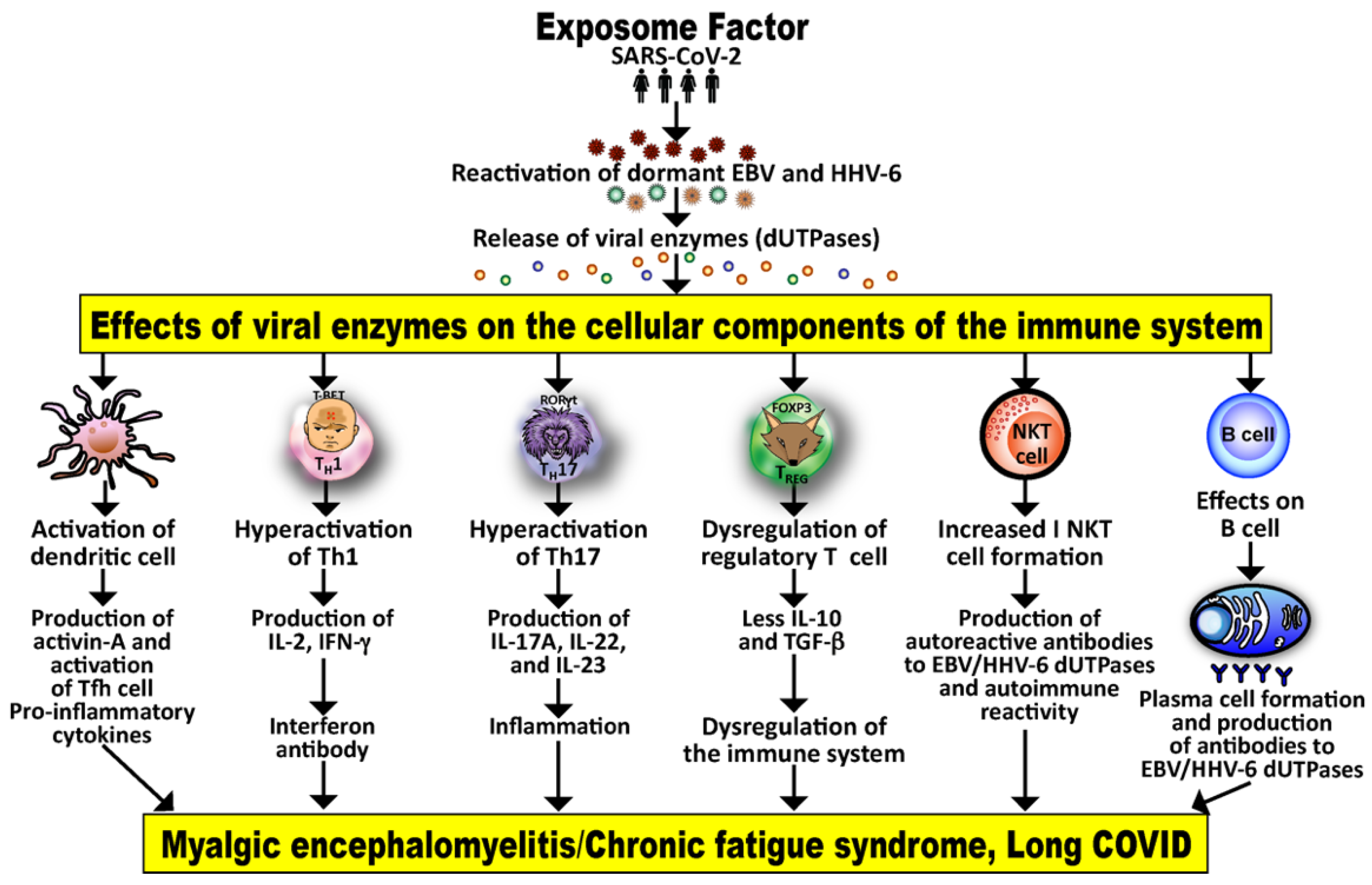
3.2. Implication of EBV and HHV-6 dUTPase in ME/CFS and Long COVID
4. Viral Superantigen Activation of the Immune System
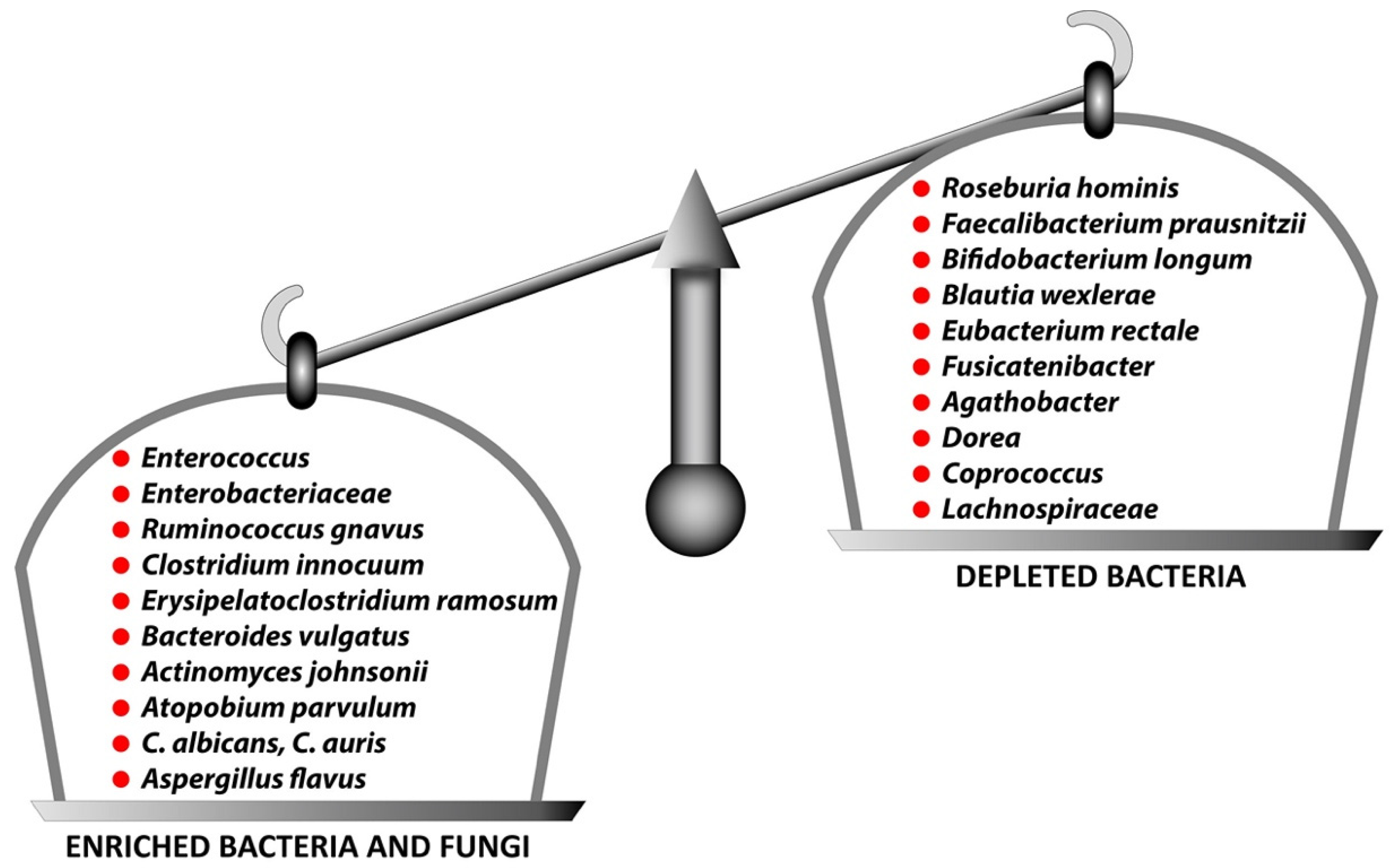
5. Disturbance in the Gut Microbiota
6. Multiple Tissue Damage and Autoimmunity
- SARS-CoV-2 spike proteins and nucleoproteins share molecular mimicry with human autoantigens involved with autoimmune diseases;
- Both animal and human monoclonal antibodies (mAbs) made against SARS-CoV-2 spike proteins and nucleoproteins react with human autoantigens;
- Antibodies made against human autoantigens react with SARS-CoV-2 spike proteins and nucleoproteins;
- The sera of patients with COVID-19 have tested positive for autoantibodies made against human autoantigens known to cross-react with SARS-CoV-2.
6.1. Herpesviruses and the Pathophysiology of Autoimmunity and Long COVID
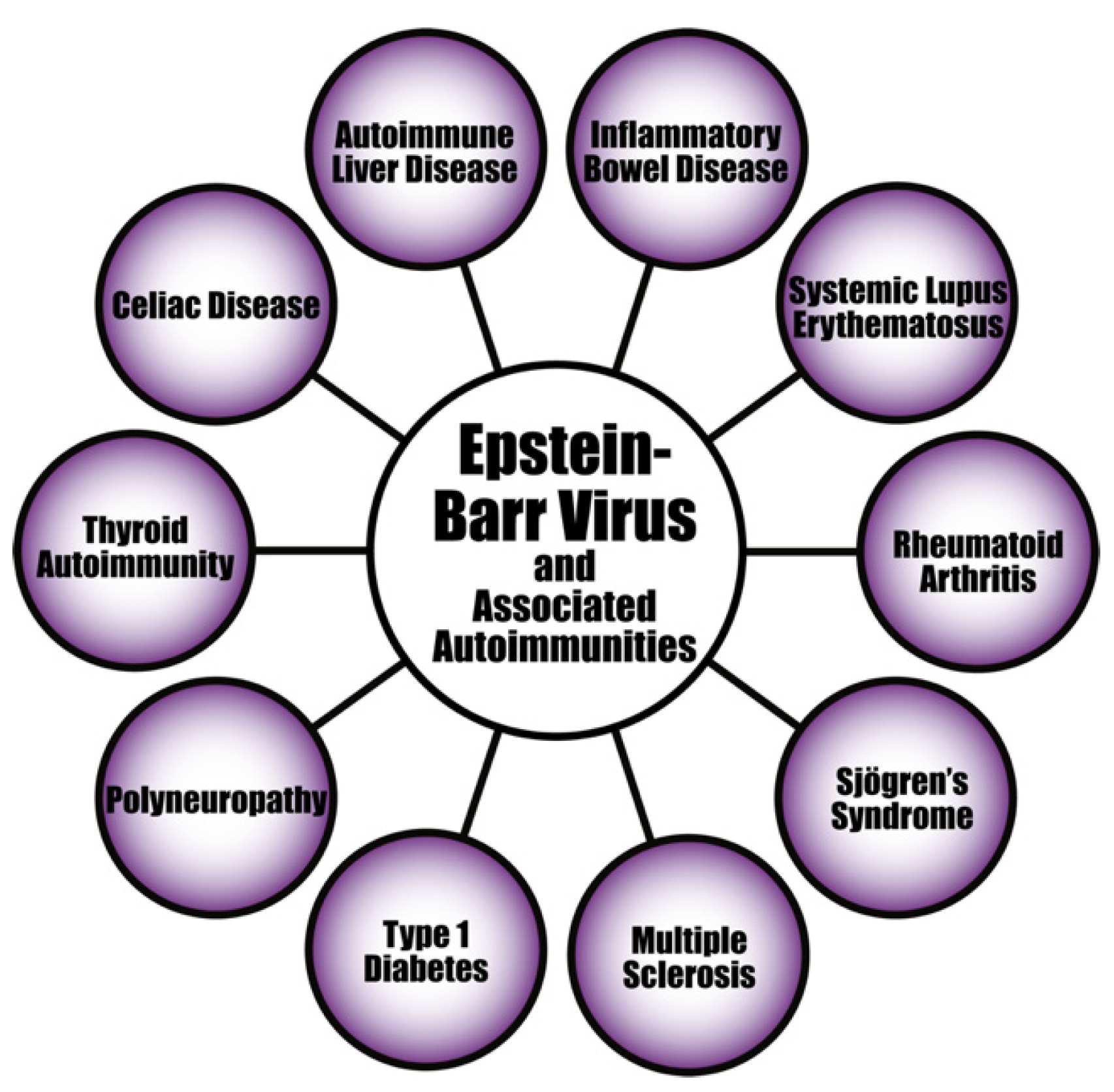
6.1.1. EBV
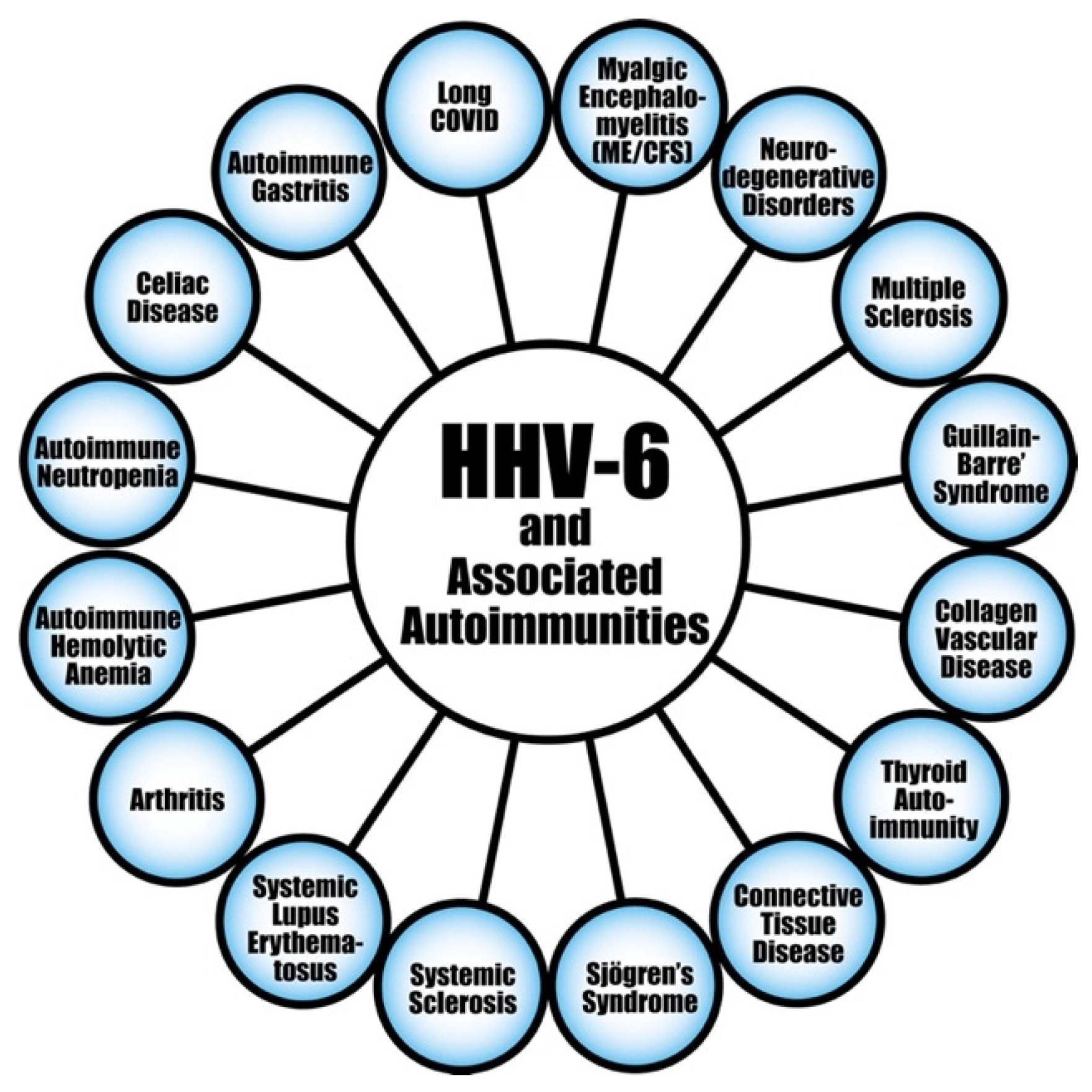
6.1.2. HHV-6A and HHV-6B
7. Treatment: Targeting Latent Viral Reactivation and Microbiota Manipulation as Strategies for the Prevention and Treatment of COVID-19 and Long COVID
8. Conclusions
- Phase 1. Acute COVID-19 with varying degrees of severity caused by viral replication and initial immune response. This can last from days to weeks, and asymptomatic patients have also been known to progress to the later phases [256].
- Phase 2. Two to five weeks after the onset of the infection, a rare hyperinflammatory condition known as multisystem inflammatory syndrome may occur, with signs and symptoms similar to Kawasaki disease. This is caused by a dysregulated immune response and can affect both children and adults [256,257,258,259].
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, Y.; Dong, Y.; Wang, L.; Xie, H.; Li, B.; Chang, C.; Wand, F.-S. Characteristics and prognostic factors of disease severity in patients with COVID-19: The Beijing experience. J. Autoimmun. 2020, 112, 102473. [Google Scholar] [CrossRef] [PubMed]
- Setiati, S.; Harimurti, K.; Safitri, E.D.; Ranakusuma, R.W.; Saldi, S.R.F.; Azwar, M.K.; Marsigit, J.; Pitoyo, Y.; Widyaningsih, W. Risk factors and laboratory test results associated with severe illness and mortality in COVID-19 patients: A systematic review. Acts Med. Indones. 2020, 52, 227–245. [Google Scholar]
- Kifer, D.; Bugada, D.; Villar-Garcia, J.; Gudelj, I.; Menni, C.; Sudre, C.; Vuckovic, F.; Ugrina, I.; Lorini, L.F.; Posso, M.; et al. Effects of environmental factors on severity and mortality of COVID-19. Front. Med. 2021, 7, 607786. [Google Scholar] [CrossRef] [PubMed]
- Vojdani, A.; Vojdani, E.; FRosenberg, A.Z.; Shoenfeld, Y. The role of exposomes in the pathophysiology of autoimmune diseases II: Pathogens. Pathophysiology 2022, 29, 243–280. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, S.M.; Smith, M.T. Epidemiology, environment and disease risks. Science 2010, 330, 460–461. [Google Scholar] [CrossRef] [Green Version]
- Vermeulen, R.; Scymanski, E.L.; Barabasi, A.L.; Miller, G.W. The exposome and health: Where chemistry meets biology. Science 2020, 367, 392–396. [Google Scholar] [CrossRef]
- Logue, J.K.; Franco, N.M.; McCulloch, D.J.; McDonald, D.; Magedson, A.; Wolf, C.R.; Chu, H.Y. Sequelae in adults at 6 months after COVID-19 infection. JAMA Netw. Open 2021, 4, e210830. [Google Scholar] [CrossRef]
- Callard, F.; Perego, E. How and why patients made long COVID. Soc. Sci. Med. 2021, 268, 113426. [Google Scholar] [CrossRef]
- Mehandru, S.; Merad, M. Pathological sequelae of long-haul COVID. Nat. Immunol. 2022, 23, 194–202. [Google Scholar] [CrossRef]
- Greenhalgh, T.; Knight, M.; A’Court, C.; Buxton, M.; Husain, L. Management of post-acute COVID-19 in primary care. BMJ 2020, 370, m3026. [Google Scholar] [CrossRef]
- Al-Jahdhami, I.; Al-Naamani, K.; Al-Mawali, A. The post-acute COVID-19 syndrome (long COVID). Oman Med. J. 2021, 36, e220. [Google Scholar] [CrossRef] [PubMed]
- Garrigues, E.; Janvier, P.; Kherabi, Y.; Le Bot, A.; Hamon, A.; Gouze, H.; Doucet, L.; Berkani, S.; Oliosi, E.; Mallart, E.; et al. Post-discharge persistent symptoms and health-related quality of life after hospitalization for COVID-19. J. Infect. 2020, 81, e4–e6. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deeks, S.G. Early clues regarding the pathogenesis of long COVID. Trends Immunol. 2022, 43, 268–270. [Google Scholar] [CrossRef]
- Al-Hadrawi, D.S.; Al-Rubaye, H.T.; Almulla, A.F.; Al-Hakeim, H.K.; Maes, M. Lowered oxygen saturation and increased body temperature in acute COVID-19 largely predict chronic fatigue syndrome and affective symptoms due to Long COVID: A precision nomothetic approach. Acta Neuropsychiatr. 2022, 22, 1–12. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G.; Ng, R.H.; Wang, K.; Choi, J.; Li, S.; Hong, S.; Zhang, R.; Xie, J.; et al. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895. [Google Scholar] [CrossRef]
- Wong, T.L.; Weitzer, D.J. Long COVID and myalgic enchepalomyelitis/chronic fatigue syndrome (MW/CFS)—A systematic review and comparison of clinical presentation and symptomatology. Medicina 2021, 57, 418. [Google Scholar] [CrossRef]
- Friedman, K.J.; Murovska, M.; Pheby, D.; Zalewski, P. Our evolving understanding of ME/CFS. Medicina 2021, 57, 200. [Google Scholar] [CrossRef]
- Pendergrast, T.; Brown, A.; Sunnquist, M.; Jantke, R.; Newton, J.L.; Strand, E.B.; Jason, L.A. Housebound versus nonhousebound patients with myalgic encephalomyelitis and chronic fatigue syndrome. Chronic Illn. 2016, 12, 292–307. [Google Scholar] [CrossRef] [Green Version]
- Chu, L.; Valencia, I.J.; Garvert, D.W.; Montoya, J.G. Onset patterns and course of myalgic encephalomyelitis/chronic fatigue syndrome. Front. Pediatr. 2019, 7, 12. [Google Scholar] [CrossRef]
- Morris, G.; Anderson, G.; Galecki, P.; Berk, M.; Maes, M. A narrative review on the similarities and dissimilarities between myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) and sickness behavior. BMC Med. 2013, 11, 64. [Google Scholar] [CrossRef] [PubMed]
- Straus, S.E.; Tosato, G.; Armstrong, G.; Lawley, T.; Preble, O.T.; Henle, W.; Davey, R.; Pearson, G.; Epstein, J.; Brus, I.; et al. Persistence illness and fatigue in adults with evidence of Epstein-Barr virus infection. Ann. Intern. Med. 1985, 102, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Kawai, K.; Kawai, A. Studies on the relationship between chronic fatigue syndrome and Epstein-Barr virus in Japan. Intern. Med. 1992, 31, 313–318. [Google Scholar] [CrossRef] [Green Version]
- Sairenji, T.; Yamanishi, K.; Tachibana, Y.; Bertoni, G.; Kurata, T. Antibody responses to Epstein-Barr virus, human herpesvirus 6 and human herpesvirus 7 in patients with chronic fatigue syndrome. Intervirology 1995, 38, 269–273. [Google Scholar] [CrossRef]
- Cameron, B.; Flamand, L.; Juwana, H.; Middledorp, J.; Naing, Z.; Rawlinson, W.; Ablashi, D.; Lloyd, A. Serological and virological investigation of the role of the herpesviruses EBV, CMV, and HHV-6 in post-infective fatigue syndrome. J. Med. Virol. 2010, 62, 1684–1688. [Google Scholar] [CrossRef]
- Cox, B.S.; Alharshawi, K.; Mena-Palomo, I.; Lafuse, W.P.; Ariza, M.E. EBV/HHV-6A dUTPases contribute to myalgic encephalomyelitis/chronic fatigue syndrome pathophysiology by enhancing Tfh cell differentiation and extrafollicular activities. JCI Insight 2022, 11, e158193. [Google Scholar] [CrossRef]
- Ariza, M.E. Myalgic encephalomyelitis/chronic fatigue syndrome: The human herpesviruses are back. Biomolecules 2021, 11, 185. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Schneider, A.M.; Mehta, A.; Sade-Feldman, M.; Kays, K.R.; Gentili, M.; Charland, N.C.; Gonye, A.L.K.; Gushterova, I.; Khanna, H.K.; et al. SARS-CoV-2 viremia is associated with distinct proteomic pathways and predicts COVID-19 outcomes. J. Clin. Investig. 2021, 131, e148635. [Google Scholar] [CrossRef] [PubMed]
- Silberry, V.G.R.; Rowe, P. Pediatric long COVID and myalgic encephalomyelitis/chronic fatigue syndrome. Ped. Inf. Dis. 2022, 41, e139–e141. [Google Scholar] [CrossRef]
- Howard-Jones, A.R.; Burgner, D.P.; Crawford, N.W.; Goeman, E.; Gray, P.E.; Hsu, P.; Kuek, S.; McMullan, B.J.; Tosif, S.; Wurzel, D.; et al. COVID-19 in children. II: Pathogenesis, disease spectrum and management. J. Pediatr. Child. Health 2022, 58, 46–53. [Google Scholar] [CrossRef]
- Proal, A.D.; VanElzakker, M.B. Long COVID or post-acute sequelae of COVID-19 (PASC): An overview of biological factors that may contribute to persistent symptoms. Front. Microbiol. 2021, 12, 698169. [Google Scholar] [CrossRef] [PubMed]
- Al-Hakeim, H.K.; Al-Rubaye, H.T.; Al-Hadrawi, D.S.; Almulla, A.F.; Maes, M. Long-COVID post-viral chronic fatigue and affective symptoms are associated with oxidative damage, lowered antioxidant defenses and inflammation: A proof of concept and mechanism study. Mol. Psychiatry 2022, 24, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Schultze, J.L.; Aschenbrenner, A.C. COVID-19 and the human innate immune system. Cell 2021, 184, 1671–1692. [Google Scholar] [CrossRef] [PubMed]
- Sumi, T.; Harada, K. Immune response to SARS-CoV-2 in severe disease and long COVID-19. iScience 2022, 25, 104723. [Google Scholar] [CrossRef]
- Griffin, D.E. Why does viral RNA sometimes persist after recovery from acute infections? PLoS Biol. 2022, 20, e3001687. [Google Scholar] [CrossRef]
- Kasuga, Y.; Zhu, B.; Jang, K.-J.; Yoo, J.-S. Innate immune sensing of coronavirus and viral evasion strategies. Exp. Mol. Med. 2021, 53, 723–736. [Google Scholar] [CrossRef]
- Li, L.; Li, S.; Pan, Y.; Qin, L.; Yang, S.; Tan, D.; Hu, Y.; Knoll, M.D.; Wang, X.; Wang, L.; et al. An immunocompetent patient with high neutralizing antibody titers who shed COVID-18 virus for 169 days—China, 2020. China CDC Wkly. 2021, 3, 688–691. [Google Scholar] [CrossRef]
- Natarajan, A.; Zlitni, S.; Brooks, E.F.; Vance, S.E.; Dahlen, A.; Hedlin, H.; Park, R.M.; Han, A.; Schmidtke, D.T.; Verma, R.; et al. Gastrointestinal symptoms and fecal shedding of SARS-CoV-2 RNA suggest prolonged gastrointestinal infection. Med 2022, 3, 371–387. [Google Scholar] [CrossRef]
- Zollner, A.; Koch, R.; Jukic, A.; Pfister, A.; Meyer, M.; Rössler, A.; Kimpel, J.; Adolph, T.E.; Tilg, H. Postacute COVID-19 is characterized by gut viral antigen persistence in inflammatory bowel diseases. Gastroenterology 2022, 163, 495–506.e8. [Google Scholar] [CrossRef]
- Rahmani, A.; Dino, G.; Leso, V.; Montecucco, A.; Vitturi, B.N.K.; Iavicoli, I.; Durando, P. Duration of SARS-CoV-2 shedding and infectivity in the working population: A systematic review and meta-analysis. Med. Lav. 2022, 113, e2022014. [Google Scholar]
- Cheung, C.C.L.; Goh, D.; Lim, X.; Tien, T.Z.; Lim, J.C.T.; Lee, J.N.; Tan, B.; Tay, Z.E.A.; Wan, W.Y.; Chen, E.X. Residual SARS-CoV-2 viral antigens detected in GI and hepatic tissues from five recovered patients with COVID-19. Gut 2022, 71, 226–229. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, J.J.L. Persistent SARS-CoV-2 infections contribute to long COVID. Med. Hypotheses 2021, 149, 110538. [Google Scholar] [CrossRef] [PubMed]
- Simonnet, A.; Engelmann, I.; Moreau, A.-S.; Garcia, B.; Six, S.; El Kalioubie, A.; Robriquet, L.; Hober, D.; Jourdain, M. High incidence of Epstein-Barr virus, cytomegalovirus and human-herpes 6 reactivation in critically ill patients with COVID-19. Infect. Dis. Now 2021, 51, 296–299. [Google Scholar] [CrossRef]
- Zubchenko, S.; Kril, I.; Nadizhko, O.; Matsyura, O.; Chopyak, V. Herpesvirus infections and post-COVID-19 manifestations: A pilot observational study. Rheumatol. Int. 2022, 42, 1523–1530. [Google Scholar] [CrossRef]
- Hoover, K.; Higginbotham, K. Epstein Barr Virus. In StatPearls; StatPearls Publishing LLC: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rasa, S.; Nora-Krukle, Z.; Henning, N.; Eliassen, E.; Shikova, E.; Harrer, T.; Scheibenbogen, C.; Murovska, M.; Prusty, B.K.; European Network on ME/CFS (EUROMENE). Chronic viral infections in myalgic encephalomyelitis/chronic fatigue syndrome {ME/CFS). J. Transl. Med. 2018, 16, 268. [Google Scholar] [CrossRef] [Green Version]
- Hadinoto, V.; Shapiro, M.; Sun, C.C.; Thorley-Lawson, D.A. The dynamics of EBV shedding implicates a central role for epithelial cells in amplifying viral output. PLoS Pathog. 2009, 5, e1000496. [Google Scholar] [CrossRef] [Green Version]
- Agut, H. Deciphering the clinical impact of acute human herpesvirus 6 (HHV-6) infections. J. Clin. Virol. 2011, 52, 164–171. [Google Scholar] [CrossRef]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternative replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar] [CrossRef]
- Aimola, G.; Beythien, G.; Aswad, A.; Kaufer, B.B. Current understanding of human herpesvirus 6 (HHV-6) chromosomal integration. Antivir. Res. 2020, 176, 104720. [Google Scholar] [CrossRef]
- Verma, D.; Church, T.M.; Swaminathan, S. Epstein-Barr virus lytic replication induxces ACE2 expression and enhances SARS-CoV-2 pseudotyped virus entry in epithelial cells. J. Virol. 2021, 95, e0019221. [Google Scholar] [CrossRef]
- Paolucci, S.; Cassaniti, I.; Novazzi, F.; Fiorina, L.; Piralla, A.; Commoli, G.; Bruno, R.; Maserati, R.; Gulminetti, R.; Novati, S.; et al. EBV DNA increase in COVID-19 patients with impaired lymphocyte subpopulation count. Int. J. Infect. Dis. 2021, 104, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of long COVID prevalence and its relationship to Epstein-Barr virus reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef]
- Balandraud, N.; Roudier, J. Epstein-Barr virus and rheumatoid arthritis. Jt. Bone Spine 2018, 85, 165–170. [Google Scholar] [CrossRef]
- Drosos, A.A.; Pelechas, E.; Voiulgari, P.V. Long COVID from rheumatology perspective: A simple mimicker or promoter of autoimmunity? Clin. Rheumatol. 2022, 41, 957–958. [Google Scholar] [CrossRef]
- Sapkota, H.R.; Nune, A. Long COVID from rheumatology perspective—A narrative review. Clin. Rheumatol. 2022, 41, 337–348. [Google Scholar] [CrossRef]
- Fan, R.; Mao, S.Q.; Gu, T.L.; Zhong, F.D.; Gong, M.L.; Hao, L.M.; Yin, F.Y.; Dong, C.Z.; Zhang, L.N. Preliminary analysis of the association between methylation of the ACE2 promoter and essential hypertension. Mol. Med. Rep. 2017, 15, 3905–3911. [Google Scholar] [CrossRef] [Green Version]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Van Sciver, N.; Lambert, P.F.; Keles, S.; et al. Epstein-Barr virus infection promotes epithelial cell growth by attenuating differentiation-dependent exit from the cell cycle. mBio 2019, 10, e01332. [Google Scholar] [CrossRef] [Green Version]
- Williams, M.V.; Cox, B.; Ariza, M.E. Herpesviruses dUTPases: A new family of pathogen-associated molecular pattern (PAMP) proteins with implications for human disease. Pathogens 2017, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Doitsh, G.; Galloway, N.L.K.; Geng, X.; Yang, Z.; Monroe, K.M.; Zependa, O.; Hunt, P.W.; Hatano, H.; Sowinski, S.; Munoz-Arias, I.; et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature 2014, 505, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.C.; Chan, J.; Clement, M.-V.; Pervaiz, S. Functional proteomics of resveratrol-induced colon cancer cell apoptosis: Caspase-6-mediated cleavage of lamin A is a major signaling loop. Proteomics 2006, 6, 2386–2394. [Google Scholar] [CrossRef]
- Wu, M.; Shen, J.; Zhan, J.; Yu, Y. dUTP pyrophosphatase, its appearance in extracellular compartment may serve as a potential biomarker for N-methyl-N’-nitro-N-nitrosoguanadine exposure in mammalian cells. Proteomics 2006, 6, 3001–3007. [Google Scholar] [CrossRef] [PubMed]
- Riza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2014, 8, e69827. [Google Scholar]
- Beutler, B. Interferences, questions and possibilities in Toll-like receptor signaling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Innate immune recognition of viral infection. Nat. Immunol. 2006, 7, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Guggemoos, S.; Hangel, D.; Hamm, S.; Heit, A.; Bauer, S.; Adler, H. TLR9 contributes to the antiviral immunity during gamma herpesvirus infection. J. Immunol. 2008, 80, 438–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Yin, H.; Zhao, M.; Lu, Q. TLR2 and TLR4 in autoimmune diseases: A comprehensive review. Clin. Rev. Allergy Immunol. 2014, 47, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual recognition of herpes simplex viruses by TLR2 and TLR9 in dendritic cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef] [Green Version]
- Boehme, K.W.; Guerrero, M.; Compton, T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J. Immunol. 2006, 177, 7094–7102. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, E.; Fiola, S.; Olivier, M.; Gosselin, J. Epstein-Barr virus induces MCP-1 secretion by human monocytes via TLR2. J. Virol. 2007, 81, 8016–8024. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.E.; Glaser, R.; Williams, M.V. Human herpesviruses-encoded dUTPases: A family of proteins that modulate dendritic cell function and innate immunity. Front. Microbiol. 2014, 5, 504. [Google Scholar] [CrossRef] [Green Version]
- Ariza, M.E.; Glaser, R.; Kaumaya, P.T.P.; Jones, C.; Williams, M. The Epstein-Barr virus (EBV)-encoded dUTPase activates NF-kappa B through the TLR2 and MyD88-dependent signaling pathway. J. Immunol. 2009, 182, 851–859. [Google Scholar] [CrossRef] [Green Version]
- Waldman, W.J.; Williams, M.V.; Lemeshow, S.A.; Binkley, P.; Guttridge, D.; Kiecolt-Glaser, J.K.; Knight, D.A.; Ladner, K.J.; Glaser, R. Epstein-Barr virus-encoded dUTPase enhances proinflammatory cytokine production by macrophages in contact with endothelial cells: Evidence for depression-induced atherosclerotic risk. Brain Behav. Immun. 2008, 22, 215–223. [Google Scholar] [CrossRef] [Green Version]
- Halpin, P.; Williams, M.V.; Klimas, N.G.; Fletcher, M.A.; Varnes, Z.; Ariza, M.E. Myalgic encephalomyelitis/chronic fatigue syndrome and Gulf War illness patients exhibit increased humoral responses to the Herpesviruses-encoded dUTPase: Implications in disease pathophysiology. J. Med. Virol. 2017, 89, 1636–1645. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.A.; Maksoud, R.; Beeraka, N.M.; Madhunapantula, S.V.; Sinelnikov, M.; Nikolenko, V.N.; Neganova, N.E.; Klochkov, S.G.; Kamal, M.A.; Staines, D.R.; et al. Analysis of postCOVID-19 condition and its overlap with myalgic encephalomyelitis/chronic fatigue syndrome. J. Adv. Res. 2022, 40, 179–196. [Google Scholar] [CrossRef] [PubMed]
- Morrow, A.K.; Malone, L.A.; Kokorelis, C.; Petracek, L.S.; Eastin, E.F.; Lobner, K.L.; Neuendorff, L.; Rowe, P.C. Long-term COVID-19 sequelae in adolescents: The overlap with orthostatic intolerance and ME/CFS. Curr. Ped. Rep. 2022, 10, 31–44. [Google Scholar] [CrossRef]
- Cui, Y.; Balshaw, D.M.; Kwok, R.K.; Thompson, C.L.; Collman, G.W.; Birnbaum, L.S. The exposome: Embracing the complexity for discovery in environmental health. Environ. Health. Perspect. 2016, 124, A137–A140. [Google Scholar] [CrossRef] [Green Version]
- Hamdy, A.; Leonardi, A. Superantigens and SARS-CoV-2. Pathogens 2022, 11, 390. [Google Scholar] [CrossRef]
- Cheng, M.H.; Zhang, S.; Porrit, R.A.; Rivas, M.N.; Pschold, L.; Willscher, E.; Binder, M.; Arditi, M.; Bahar, I. Superantigenic character of an insert unique to SARS-CoV-2 spike supported by skewed TCR repertoire in patients with hyperinflammation. Prot. Nat. Acad. Sci. USA 2020, 117, 25254–25262. [Google Scholar] [CrossRef]
- Scaglioni, V.; Soriano, E.R. Are superantigens the cause of cytokine storm and viral sepsis in severe COVID-19? Observations and hypothesis. Scand. J. Immunol. 2020, 92, e12944. [Google Scholar] [CrossRef]
- Gammazza, A.M.; Légaré, S.; Lo Bosco, G.; Fucarino, A.; Angileri, F.; de Macario, E.C.; Macario, A.J.L.; Cappello, F. Human molecular chaperones share with SARS-CoV-2 antigenic epitopes potentially capable of eliciting autoimmunity against endothelial cells: Possible role of molecular mimicry in COVID-19. Cell Stress Chaperones 2020, 25, 737–741. [Google Scholar] [CrossRef]
- Lubkowska, A.; Pluta, W.; Stronska, A.; Lalko, A. Role of heat shock proteins (Hsp70 and Hsp90) in viral infection. Int. J. Mol. Sci. 2021, 22, 9366. [Google Scholar] [CrossRef] [PubMed]
- Lanneau, D.; Brunet, M.; Frisan, E.; Solary, E.; Fontenay, M.; Garrido, C. Heat shock proteins: Essential proteins for apoptosis regulation: Apoptosis Review Series. J. Cell. Mol. Med. 2008, 12, 743–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, F.H.N.; Kwan, A.; Winder, N.; Mughal, A.; Collado-Rojas, C.; Muthana, M. Understanding immune response to viruses—Do underlying Th1/Th2 cell biases predict outcome? Viruses 2022, 14, 1493. [Google Scholar] [CrossRef] [PubMed]
- Nagler, C.R. Modern World Influences on the Microbiome and Their Consequences for Immune-Mediated Disease. J. Immunol. 2021, 207, 1695–1696. [Google Scholar] [CrossRef]
- Blaser, M. The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol. 2017, 17, 461–463. [Google Scholar] [CrossRef]
- Khan, M.F.; Wang, G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2017, 7, 22–27. [Google Scholar] [CrossRef]
- Dehner, C.; Fine, R.; Kriegel, M. The microbiome in systemic autoimmune disease: Mechanistic insights from recent studies. Curr. Opin. Rheumatol. 2019, 31, 201–207. [Google Scholar] [CrossRef]
- De Luca, F.; Shoenfeld, Y. The microbiome in autoimmune diseases. Clin. Exp. Immunol. 2018, 195, 74–85. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Selmi, C.; Tang, R.; Gershwin, M.E.; Ma, X. The microbiome and autoimmunity: A paradigm from the gut–liver axis. Cell. Mol. Immunol. 2018, 15, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Dotan, A.; Mahroum, N.; Bogdanos, D.P.; Shoenfeld, Y. COVID-19 as an infectome paradigm of autoimmunity. J. Allergy Clin. Immunol. 2022, 149, 63–64. [Google Scholar] [CrossRef]
- Wang, H.; Wang, H.; Sun, Y.; Ren, Z.; Zhu, W.; Li, A.; Cui, G. Potential associations between microbiome and COVID-19. Front. Med. 2021, 8, 785496. [Google Scholar] [CrossRef] [PubMed]
- Yeoh, Y.K. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Reinold, J.; Farahpour, F.; Schoerding, A.-K.; Fehring, C.; Dolff, S.; Konik, M.; Korth, J.; van Baal, L.; Buer, J.; Witzke, O.; et al. The fungal gut microbiome exhibits reduced diversity and increased relative abundance of Ascomycota in severe COVID illness and distinct interconnected communities in SARS-CoV-2 positive patients. Front. Cell. Infect. Microbiol. 2022, 12, 848650. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Zhan, H.; Zhang, F.; Liu, Q.; Tso, E.Y.K.; Lui, G.C.Y.; Chen, N.; Li, A.; Lu, W.; Chan, F.K.L.; et al. Alterations in fecal fungal microbiome of patients with covid-19 during time of hospitalization until discharge. Gastroenterology 2020, 159, 1302–1310. [Google Scholar] [CrossRef]
- Liu, Q.; Mak, J.W.Y.; Su, Q.; Yeoh, Y.K.; Lui, G.C.Y.; Ng, S.S.S.; Zhang, F.; Li, A.Y.L.; Lu, W.; Hui, D.S.C.; et al. Gut microbiota dynamics in a prospective cohort of patients with post-acute COVID-19 syndrome. Gut 2022, 71, 544–552. [Google Scholar] [CrossRef]
- Zhang, F.; Wan, Y.; Zuo, T.; Yeoh, Y.K.; Liu, Q.; Zhang, L.; Zhan, H.; Lu, W.; Xu, W.; Lui, G.C.Y.; et al. Prolonged impairment of short-chain fatty acid and l-isoleucine biosynthesis in gut microbiome in patients with COVID-19. Gastroenterology 2022, 162, 548–561. [Google Scholar] [CrossRef]
- Venzon, M.; Cadwell, K. COVID-19 and the forgotten organ: Prolonged changes to the metabolic ouptut of the gut microbiome. Gastroenterology 2022, 162, 394–396. [Google Scholar] [CrossRef]
- Zuo, T.; Liu, Q.; Zhang, F.; Lui, G.C.-Y.; Tso, E.Y.; Yeoh, Y.K.; Chen, Z.; Boon, S.S.; Chan, F.K.; Chan, P.K.; et al. Depicting SARS-CoV-2 faecal viral activity in association with gut microbiota composition in patients with COVID-19. Gut 2021, 70, 276–284. [Google Scholar] [CrossRef]
- Hejrati, A.; Rafiei, A.; Soltanshahi, M.; Hosseinzadeh, S.; DAbiri, M.; Taghadosi, M.; Taghiloo, S.; Bashash, D.; Khorsidi, F.; Zafari, P. Innate immune response in systemic autoimmune diseases: A potential target of therapy. Inflammopharmacology 2020, 28, 1421–1438. [Google Scholar] [CrossRef]
- Bordin, P.; Casari, G.; Townsend, L.; O’Farrelly, C.; Tancevski, I.; Löffler-Ragg, J.; Mogensen, T.H.; Casanova, J.L.; COVID Human Genetic Effort. Studying severe long COVID to understand post-infectious disorders beyond COVID-19. Nat. Med. 2022, 28, 879–889. [Google Scholar] [CrossRef]
- Vojdani, A.; Kharrazian, D. Potential antigenic cross-reactivity between SARS-CoV-2 and human tissue with a possible link to an increase in autoimmune diseases. Clin. Immunol. 2020, 217, 108480. [Google Scholar] [CrossRef]
- Vojdani, A.; Vojdani, E.; Melgar, A.L.; Redd, J. Reaction of SARS-CoV-2 antibodies with other pathogens, vaccines, and food antigens. Front. Immunol. 2022, 13, 1003094. [Google Scholar] [CrossRef]
- Churilov, L.P.; Normatov, M.G.; Utekhin, V.J. Molecular mimicry between SARS-CoV-2 and human endocrinocytes: A prerequisite of post-COVID-19 endocrine autoimmunity? Pathophysiology 2022, 29, 486–494. [Google Scholar] [CrossRef]
- Ormiston, C.K.; Swiatkiewicz, I.; Taub, P.R. Postural orthostatic syndrome as a sequela of COVID-19. Heart Rhythm 2022, 19, 1880–1889. [Google Scholar] [CrossRef]
- Cabral-Marques, O.; Halpert, G.; Schimke, L.F.; Ostrinski, Y.; Vojdani, A.; Baiocchi, G.C.; Freire, P.P.; Filgueiras, I.S.; Zyskind, I.; Lattin, M.T.; et al. Autoantibodies targeting GPCRs and RAS-related molecules associate with COVID-19 severity. Nat. Commun. 2022, 13, 1220. [Google Scholar] [CrossRef]
- Sancho-Shimizu, V.; Brodin, P.; Cobat, A.; Biggs, C.M.; Toubiana, J.; Lucas, C.L.; Henrickson, S.E.; Belot, A.; MIS-C@CHGE; Tangye, S.G. SARS-CoV-2–related MIS-C: A key to the viral and genetic causes of Kawasaki disease? J. Exp. Med. 2021, 218, e20210446. [Google Scholar] [CrossRef]
- Consiglio, C.R.; Cotugno, N.; Sardh, F.; Pou, C.; Amodio, D.; Rodriguez, L.; Tan, Z.; Zicari, S.; Ruggiero, A.; Pascucci, G.R.; et al. The immunology of multisystem inflammatory syndrome in children with COVID-19. Cell 2020, 183, 968–981. [Google Scholar] [CrossRef]
- Novelli, L.; Motta, F.; De Santis, M.; Ansari, A.A.; Gershwin, M.E.; Selmi, C. The JANUS of chronic inflammatory and autoimmune diseases onset during COVID-19—A systematic review of the literature. J. Autoimmun. 2021, 117, 102592. [Google Scholar] [CrossRef]
- Rodriguez, Y.; Novelli, L.; Rojas, M.; De Santis, M.; Acosta-Ampudia, Y.; Monsalve, D.M.; Ramirez-Santana, C.; Cistanzo, A.; Ridgway, W.M.; Ansari, A.A.; et al. Autoinflammatory and autoimmune conditions at the crossroad of COVID-19. J. Autoimmun. 2020, 114, 102506. [Google Scholar] [CrossRef]
- Gianini, M.; Ohana, M.; Nespola, B.; Zanframundo, G.; Geny, B.; Meyer, A. Similarities between COVID-19 and anti-MDA5 syndrome: What can we learn for better care? Eur. Respir. J. 2020, 56, 2001618. [Google Scholar] [CrossRef]
- Wang, Y.; Du, G.; Zhang, G.; Matucci-Cerinic, M.; Furst, D.E. Similarities and differences between severe COVID-19 pneumonia and MDA-5-positive dermatomyositis-associated rapidly progressive interstitial lung diseases: A challenge for the future. Ann. Rheum. Dis. 2022, 81, e192. [Google Scholar] [CrossRef]
- Zhang, Q.; Bastard, P.; Liu, Z.; Le Pen, J.; Moncada-Velez, M.; Chen, J.; Ogishi, M.; Sabli, I.K.D.; Hodeib, S.; Korol, C.; et al. Inborn errors of type 1 IFN immunity in patients with life-threatening COVID-19. Science 2020, 370, eabd4570. [Google Scholar] [CrossRef]
- Bastard, P.; Rosen, L.B.; Zhang, Q.; Michailidis, E.; Hoffmann, H.-H.; Zhang, Y.; Dorgham, K.; Philippot, Q.; Rosain, J.; Béziat, V.; et al. Autoantibodies against type1 IFNs in patients with life-threatening COVID-19. Science 2020, 370, eabd4585. [Google Scholar] [CrossRef]
- De Stefano, L.; Rossi, S.; Montecucco, C.; Bugatti, S. Transient monoarthritis and psoriatic skin lesions following COVID-19. Ann. Rheum. Dis. 2020, 2020, 218520. [Google Scholar] [CrossRef]
- Liew, I.Y.; Mak, T.M.; Cui, L.; Vasoo, S.; Lim, X.R. A case of reactive arthritis secondary to Coronavirus Disease 2019 infection. J. Clin. Rheumatol. 2020, 26, 233. [Google Scholar] [CrossRef]
- Ono, K.; Kishimoto, M.; Shimasaki, T.; Uchida, H.; Kurai, D.; Deshpande, G.A.; Komagata, Y.; Kaname, S. Reactive arthritis after COVID-19 infection. R.M.D. Open 2020, 6, e001350. [Google Scholar] [CrossRef]
- Novelli, L.; Motta, F.; Ceribelli, A.; Guidelli, G.M.; Luciano, N.; Isailovic, N.; Vecellio, M.; Caprioli, M.; Clementi, N.; Clementi, M.; et al. A case of psoriatic arthritis triggered by SARS-CoV-2 infection. Rheumatology 2021, 60, e21–e23. [Google Scholar] [CrossRef]
- Talarico, R.; Stagnaro, C.; Ferro, F.; Carli, L.; Mosca, M. Symmetric peripheral polyarthritis developed during SARS-CoV-2 infection. Lancet Rheumatol. 2020, 2, e518–e519. [Google Scholar] [CrossRef]
- Saricaoglu, E.M.; Hasanoglu, I.; Guner, R. The first reactive arthritis case associated with COVID-19. J. Med. Virol. 2021, 93, 192–193. [Google Scholar] [CrossRef]
- Yokogawa, N.; Minematsu, M.; Katano, H.; Suzuki, T. Case of acute arthritis following SARS-CoV-2 infection. Ann. Rheum. Dis. 2021, 80, e101. [Google Scholar] [CrossRef]
- Zuo, Y.; Estes, S.K.; Ali, R.A.; Gandhi, A.A.; Yalavarthi, S.; Shi, H.; Sule, G.; Gockma, K.; Madison, J.A.; Zuo, M.; et al. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zuo, Y.; Navaz, S.; Harbaugh, A.; Hoy, C.K.; Gandhi, A.A.; Sule, G.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; et al. Endothelial cell-activating antibodies in COVID-19. medRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ramos, A.E.; Martin-Nares, E.; Hernández-Molina, G. New onset of autoimmune diseases following COVID-19 diagnosis. Cells 2021, 10, 3592. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, P.; Fallahi, M.S.; Erabi, G.; Pakdin, M.; Zarezadeh, S.M.; Faridzadeh, A.; Entezari, S.; Ansari, A.; Poudineh, M.; Deravi, N. Multisystem inflammatory syndrome and autoimmune disease following COVID-19: Molecular mechanisms and therapeutic opportunities. Front. Mol. Biosci. 2022, 9, 804109. [Google Scholar] [CrossRef] [PubMed]
- Halpert, G.; Shoenfeld, Y. SARS-CoV-2, the Autoimmune virus. Autoimmun. Rev. 2020, 19, 102695. [Google Scholar] [CrossRef]
- Dotan, A.; Muller, S.; Kanduc, D.; David, P.; Halpert, G.; Shoenfeld, Y. The SARS-CoV-2 as an instrumental trigger of autoimmunity. Autoimmun. Rev. 2021, 20, 102792. [Google Scholar] [CrossRef]
- Shoenfeld, Y. Corona (COVID-19) time musings: Our involvement in COVID-19 pathogenesis, diagnosis, treatment and vaccine planning. Autoimmune Res. 2020, 19, 102538. [Google Scholar] [CrossRef] [PubMed]
- Lyons-Weiler, J. Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity. J. Transl. Autoimmun. 2020, 3, 100051. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damorakin, G.; Gkavogianni, T.; Adami, M.-E.; Katsaounou, P.; et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar]
- Vojdani, A.; Vojdani, E.; Kharrazian, D. Reaction of human monoclonal antibodies to SARS-CoV-2 proteins with tissue antigens: Implications for autoimmune diseases. Front. Immunol. 2021, 11, 617089. [Google Scholar] [CrossRef]
- Kanduc, D.; Shoenfeld, Y. On the molecular determinants of the SARS-CoV-2 attack. Clin. Immunol. 2020, 215, 108426. [Google Scholar] [CrossRef] [PubMed]
- Kanduc, D.; Shoenfeld, Y. Molecular mimicry between SARS-CoV-2 spike glycoprotein and mammalian proteomes: Implications for the vaccine. Immunol. Res. 2020, 68, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Becker, R.C. COVID-19-associated coagulopathy. J. Thromb. Thrombolysis 2020, 50, 54–67. [Google Scholar] [PubMed]
- Zhang, Y.; Xiao, M.; Zhang, S.; Xia, P.; Cao, W.; Jiang, W.; Chen, H.; Ding, X.; Zhao, H.; Zhang, H.; et al. Coagulopathy and antiphospholipid antibodies in patients with COVID-19. N. Engl. J. Med. 2020, 382, e38. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sawalha, A.H.; Lu, Q. COVID-19 and autoimmune diseases. Curr. Opin. Rheumatol. 2020, 33, 155–162. [Google Scholar]
- Ehrenfeld, M.; Tincani, A.; Andreoli, L.; Cattalini, M.; Greenbaum, A.; Kanduc, D.; Alijotas-Reig, J.; Zinserling, V.; Semenova, N.; Amital, H.; et al. COVID-19 and autoimmunity. Autoimmun. Rev. 2020, 19, 102597. [Google Scholar]
- Ryabkova, V.A.; Churilov, L.P.; Shoenfeld, Y. Influenza infection, SARS, MERS and COVID-19: Cytokine storm—The common denominator and the lessons to be learned. Clin. Immunol. 2020, 223, 108652. [Google Scholar]
- Malkova, A.; Kudlay, D.; Kudryavtsev, I.; Starshinova, A.; Yablonskiy, P.; Shoenfeld, Y. Immunogenetic predictors of severe COVID-19. Vaccines 2021, 9, 211. [Google Scholar] [CrossRef]
- Mahroum, N.; Alghory, A.; Kiyak, Z.; Alwani, A.; Seida, R.; Alrais, M.; Shoenfeld, Y. Ferritin—From iron, through inflammation and autoimmunity, to COVID-19. J. Autoimmun. 2021, 126, 102778. [Google Scholar]
- Vojdani, A. Molecular and immunological evidence for SARS-CoV-2 being the autoimmune virus. In Proceedings of the Abstract, Oral Presentation, 13th International Congress on Autoimmunity, Athens, Greece, 10–13 June 2022. [Google Scholar]
- Nahmias, A.J. Clinical aspects of infection with herpes simplex viruses 1 and 2. In The Human Herpesviruses: An Interdisciplinary Perspective; Nahmias, A., Dowdle, W.R., Shinazai, R.F., Eds.; Elsevier: Amsterdam, The Netherlands, 1981. [Google Scholar]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. C.N.S. Neurol. Disord.-Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [Green Version]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 370516. [Google Scholar]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 535738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, S.-F.; Wu, H.-C.; Tsai, W.-C.; Yen, J.-H.; Chiang, W.; You, C.-Y.; Lu, S.-N.; Chiang, L.-C.; Chen, C.-J. Detecting Epstein-Barr virus DNA from peripheral blood mononuclear cells in adult patients with systemic lupus erythematosus in Taiwan. Med. Microbiol. Immunol. 2004, 194, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Moon, U.Y.; Park, S.J.; Oh, S.T.; Kim, W.-U.; Park, S.-H.; Lee, C.-H.; Cho, C.-S.; Kim, H.-Y.; Lee, W.-K. Patients with systemic lupus erythematosus have abnormally elevated Epstein-Barr virus load in blood. Arthritis Res. Ther. 2004, 6, R295–R302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, A.J.; Hochberg, D.; Rand, W.M.; Thorley-Lawson, D.A. EBV and systemic lupus erythematosus: A new perspective. J. Immunol. 2005, 174, 6599–6607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draborg, A.H.; Jorgensen, J.M.; Müller, H.; Nielsen, C.T.; Jacobsen, S.; Iversen, L.V.; Theander, E.; Nielsen, L.P.; Houen, G.; Duus, K. Epstein-Barr virus early antigen diffuse (EBV-EA/D)-directed immunoglobulin A antibodies in systemic lupus erythematosus patients. Scand. J. Rheumatol. 2012, 41, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr-virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar]
- Jog, N.R. Association of Epstein-Barr virus serological reactivation with transitioning to systemic lupus erythematosus in at-risk individuals. Ann. Rheum. Dis. 2019, 78, 1235–1241. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J. High prevalence of immunoglobulin A antibody against Epstein-Barr virus capsid antigen in adult patients with lupus with disease flare: Case control studies. J. Rheumatol. 2005, 32, 44–47. [Google Scholar]
- Kaufman, K.M. Peptide mimics of a major lupus epitope of SmB/B′. Ann. N. Y. Acad. Sci. 2003, 987, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Mclain, M.T. An altered immune response to Epstein-Barr nuclear antigen 1 in pediatric systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 360–368. [Google Scholar]
- Poole, B.D. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J Autoimmun. 2008, 31, 362371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandman-Goddard, G. Exposure to Epstein-Barr virus infection is associated with mild systemic lupus erythematosus disease. Ann. N. Y. Acad. Sci. 2009, 1173, 658–663. [Google Scholar] [CrossRef]
- Esen, B.A. Serologic response to Epstein-Barr virus antigens in patients with systemic lupus erythematosus: A controlled study. Rheumatol. Int. 2012, 32, 79–83. [Google Scholar] [CrossRef]
- Vista, E.S. Strong viral associations with SLE among Filipinos. Lupus Sci. Med. 2017, 4, e000214. [Google Scholar]
- Morissette, G.; Flamand, L. Herpesviruses and chromosomal integration. J. Virol. 2010, 84, 12100–12109. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Lafuente, R.; Fernández-Gutiérrez, B.; de Miguel, S.; Jover, J.A.; Rollin, R.; Loza, E.; Clemente, D.; Lamas, J.R. Potential relationship between herpes viruses and rheumatoid arthritis: Analysis with qualitative real time polymerase chain reaction. Ann. Rheum. Dis. 2005, 64, 1357–1359. [Google Scholar] [CrossRef] [Green Version]
- Krueger, G.R.F.; Sander, C.; Hoffmann, A.; Barth, A.; Koch, B.; Braun, M. Isolation of human herpesvirus-6 (HHV-6) from patients with collagen vascular diseases. In Vivo 1991, 5, 217–225. [Google Scholar]
- Hoffman, A.; Kirn, E.; Kuerten, A.; Sander, C.; Krueger, G.R.F.; Ablashi, D.V. Active human herpesvirus-6 (HHV-6) infection associated with Kikuchi-Fujimoto disease and systemic lupus erythematosus (SLE). In Vivo 1991, 5, 265–269. [Google Scholar]
- Broccolo, F.; Drago, F.; Paolino, S.; Cassina, G.; Gatto, F.; Fusetti, L.; Matteoli, B.; Zaccaria, E.; Parodi, A.; Lusso, P.; et al. Reactivation of human herpesvirus 6 (HHV-6) infection in patients with connective tissue diseases. J. Clin. Virol. 2009, 46, 43–46. [Google Scholar] [PubMed]
- Broccolo, F.; Drago, F.; Cassina, G.; Fava, A.; Fusetti, L.; Matteoli, B.; Ceccherini-Nelli, L.; Sabbadini, M.G.; Lusso, P.; Parodi, A.; et al. Selective reactivation of human herpesvirus 6 in patients with autoimmune connective tissue diseases. J. Med. Virol. 2013, 85, 1925–1934. [Google Scholar] [PubMed]
- Ranger-Rogez, S.; Vidal, E.; Liozon, F.; Denis, F. Primary Sjögren’s syndrome and antibodies to human herpesvirus type 6. Clin. Infect. Dis. 1994, 19, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Soffritti, I.; D’Accolti, M.; Bortolotti, D.; Sighinolfi, G.; Giuggioli, D.; Ferri, C. HHV-6A infection and systemic sclerosis: Clues of a possible association. Microorganisms 2020, 8, 39. [Google Scholar] [CrossRef] [Green Version]
- Caselli, E.; Zatelli, M.C.; Rizzo, R.; Benedetti, S.; Martorelli, D.; Trasforini, G.; Cassai, E.; degli Uberti, E.C.; Di Luca, D.; Dolcetti, R. Virologic and immunologic evidence supporting an association between HHV-6 and Hashimoto’s thyroiditis. PLoS Pathog. 2012, 8, 100295. [Google Scholar]
- Sultanova, A.; Cistjakovs, M.; Gravelsina, S.; Chapenko, S.; Roga, S.; Cunskis, E.; Nore-Krukle, Z.; Groma, V.; Ventina, I.; Murovska, M. Association of active human herpesvirus 6 (HHV-6) infection with autoimmune thyroid gland diseases. Clin. Microbiol. Infect. 2017, 23, e1–e50. [Google Scholar] [CrossRef]
- Soldan, S.S.; Berti, R.; Salem, N.; Secchiero, P.; Flamand, L.; Calabresi, P.A.; Brennan, M.B.; Maloni, H.W.; McFarland, H.F.; Lin, H.C.; et al. Association of human herpesvirus 6 (HHV-6) with multiple sclerosis: Increased IgM response to HHV-6 early antigen and detection of serum HHV-6 DNA. Nature Med. 1997, 3, 1394–1397. [Google Scholar] [CrossRef]
- Fillet, A.M.; Lozeron, P.; Augt, H.; Lyon-Caen, O.; Liblau, R. HHV-6 and multiple sclerosis. Nat. Med. 1998, 4, 537. [Google Scholar] [CrossRef]
- Coates, A.R.; Bell, J. HHV-6 and multiple sclerosis. Nat. Med. 1998, 4, 537–538. [Google Scholar] [CrossRef]
- Ablashi, D.V.; Lapps, W.; Kaplan, M.; Whitman, J.E.; Richert, J.R.; Pearson, G.R. Human herpesvirus-6 (HHV-6) in multiple sclerosis: A preliminary report. Mult. Scler. 1998, 4, 490–496. [Google Scholar] [CrossRef]
- Friedman, J.E.; Lyons, M.J.; Cu, G.; Ablashi, D.V.; Whitman, J.E.; Edgar, M.; Koskiniemi, M.; Vaheri, A.; Zabriskie, J.B. The association of the human herpesvirus-6 and MS. Mult. Scler. 1999, 5, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Voumvourakis, K.I.; Kitsos, D.K.; Tsiodras, S.; Petrikkos, G.; Stamboulis, E. Human herpesvirus 6 infection as a trigger of multiple sclerosis. Mayo Clin. Proc. 2010, 11, 1023–1030. [Google Scholar]
- Akhyani, N.; Berti, R.; Brennan, M.B.; Soldan, S.S.; Eaton, J.M.; McFarland, H.F.; Jacobson, S. Tissue distribution and variant characterization of human herpesvirus (HHV-6) increased prevalence of HHV-6A in patients with multiple sclerosis. J. Infect. Dis. 2000, 182, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Sanders, V.J.; Felisan, S.; Waddell, A.; Tourtellotte, W.W. Detection of Herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J. Neurovirol. 1996, 2, 249–258. [Google Scholar] [CrossRef]
- Kim, J.-S.; Lee, K.-S.; Park, J.-H.; Kim, M.-Y.; Shin, W.-S. Detection of human herpesvirus 6 variant A in peripheral blood mononuclear cells from multiple sclerosis patients. Eur. Neurol. 2000, 43, 170–173. [Google Scholar] [CrossRef]
- Chapenko, S.; Millers, A.; Nora, Z.; Logina, I.; Kukaine, R.; Murovska, M. Correlation beteeen HHV-6 reactivation and multiple sclerosis disease activity. J. Med. Virol. 2003, 69, 111–117. [Google Scholar] [CrossRef]
- Rotola, A.; Merlotti, I.; Caniatti, L.; Caselli, E.; Granieri, E.; Tola, M.R.; Di Luca, D.; Cassai, E. Human herpesvirus 6 infects the central nervous system of multiple sclerosis patients in the early stages of the disease. Mult. Scler. 2004, 10, 348–354. [Google Scholar] [CrossRef]
- Goodman, A.D.; Mock, D.J.; Powers, J.M.; Baker, J.V.; Blumberg, B.M. Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J. Infect. Dis. 2003, 187, 1365–1376. [Google Scholar] [CrossRef] [Green Version]
- Wilborn, F.; Schmidt, C.A.; Brinckmann, V.; Jendroska, K.; Oettle, H.; Siegert, W. A potential role for human herpesvirus type 6 in nervous system disease. J. Neuroimmunol. 1994, 49, 213–214. [Google Scholar] [CrossRef]
- Alvarez-Lafuente, R.; De Las Heras, V.; Bartolome, M.; Picazo, J.J.; Arroyo, R. Relapsing-remitting multiple sclerosis and human herpesvirus 6 active infection. Arch. Neurol. 2004, 61, 1523–1527. [Google Scholar] [CrossRef] [Green Version]
- Simpson, S.; Taylor, B.; Dwyer, D.E.; Taylor, J.; Blizzard, L.; Ponsonby, A.-L.; Pittas, F.; Dwyer, T.; van der Mei, I. Anti-HHV-6 IgG titer significantly predicts subsequent relapse risk in multiple sclerosis. Mult. Scler. 2012, 18, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, S.; Dominguez-Mozo, M.; García-Martinez, M.A.; Garcia-Frontini, M.C.; Villarubia, N.; Costa-Frossard, L.; Villar, L.M.; Arroyo, R.; Alvarez-Lafuente, R. Anti-human herpesvirus 6 A/B antibodies titers correlate with multiple sclerosis-associated retrovirus envelope expression. Front. Immunol. 2021, 12, 798003. [Google Scholar] [CrossRef]
- Dominguez-Mozo, M.I.; Perez-Perez, S.; Villarubia, N.; Costa-Frossard, L.; Fernandez-Velasco, J.I.; Ortega-Madueño, I.; Garcia-Martinez, M.A.; Garcia-Calvo, E.; Estevez, H.; Garcia, J.L.L.; et al. Herpesvirus antibodies, vitamin D and short-chain fatty acids: Their correlation with cell subsets in multiple sclerosis patients and healthy controls. Cells 2021, 10, 119. [Google Scholar] [CrossRef] [PubMed]
- Grima, P.; Chiavaroli, R.; Calabrese, P.; Tundo, P. Severe hepatitis with autoimmune features following a HHV-6: A case report. Cases J. 2008, 1, 110. [Google Scholar] [CrossRef] [Green Version]
- Yagasaki, H.; Kato, M.; Shimizu, N.; Shichino, H.; Chin, M.; Mugishima, H. Autoimmune hemolytic anemia and autoimmune neutropenia in a child with erythroblastopenia of childhood (TEC) caused by human herpesvirus-6 (HHV-6). Ann. Hematol. 2011, 90, 851. [Google Scholar] [CrossRef]
- Faierstein, K.; Shilo, N.; Levartovsky, A.; Raphael, R.; Givon, A.; Agmon-Levin, N.; Mayan, H. Autoimmune neutropenia associated with HHV-6 virus infection: A case report. Front. Immunol. 2022, 13, 880016. [Google Scholar] [CrossRef]
- Chapenko, S.; Krumina, A.; Kozireva, S.; Nora, Z.; Sultanova, A.; Viksna, L.; Murovska, M. Activation of human herpesviruses 6 and 7 in patients with chronic fatigue syndrome. J. Clin. Virol. 2006, 37 (Suppl. S1), S47–S51. [Google Scholar] [CrossRef]
- Chapenko, S.; Krumina, A.; Logina, I.; Rasa, S.; Chistjakovs, M.; Sultanova, A.; Viksna, L.; Murovska, M. Association of active human herpesvirus-6, -7 and parvovirus b19 infection with clinical outcomes in patients with myalgic encephalomyelitis/chronic fatigue syndrome. Adv. Virol. 2012, 2012, 205085. [Google Scholar] [CrossRef] [Green Version]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfuncrtion. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar]
- Morris, G.; Maes, M. Mitochondrial dysfunctions in myalgic encephalomyelitis/chronic fatigue syndrome explained by activated immuno-inflammatory, oxidative and nitrosative stress pathways. Metab. Brain Dis. 2014, 29, 19–36. [Google Scholar] [CrossRef]
- Schreiner, P.; Harrer, T.; Scheibenbogen, C.; Lamer, S.; Schlosser, A.; Naviaux, R.K.; Prusty, B.K. Human herpesvirus-6 reactivation, mitochondrial fragmentation, and the coordination of antiviral and metabolic phenotypes in myalgic encephalomyelitis/chronic fatigue syndrome. Immunohorizons 2020, 4, 201–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lino, K.; Alves, L.S.; Raposo, J.V.; Medeiros, T.; Souza, C.F.; da Silva, A.A.; de Paula, V.S.; Almeida, J.R. Presence and clinical impact of human herpesvirus-6 infection in patients with moderate to critical coronavirus disease-19. J. Med. Virol. 2022, 94, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Drago, F.; Ciccarese, G.; Rebora, A.; Parodi, A. Human herpesvirus-6, -7, and Epstein-Barr virus reactivation inpityriasis roseaduring COVID-19. J. Med. Virol. 2020, 93, 1850–1851. [Google Scholar] [CrossRef] [PubMed]
- Bigley, T.M.; Yang, L.; Kang, L.-I.; Saenz, J.B.; Victorino, F.; Yokoyama, W.M. Disruption of thymic central tolerance by infection with murine roseolovirus induces autoimmune gastritis. J. Exp. Med. 2022, 219, e20211403. [Google Scholar] [CrossRef]
- Rojas, M.; Rodriguez, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Zhu, C.; Li, Q.-Z.; Ramirez-Santana, C.; Anaya, J.-M. Autoimmunity is a hallmark of post-COVID syndrome. J. Transl. Med. 2022, 20, 129. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B.; Howe, A. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef]
- Derwand, R.; Scholz, M.; Zelenko, V. COVID-19 outpatients: Early risk-stratified treatment with zinc plus low-dose hydroxychloroquine and azythromycin. A retrospective case series study. Int. J. Antimicrob. Agents 2020, 56, 106214. [Google Scholar] [CrossRef]
- Formiga, F.R.; Leblanc, R.; Reboucas, J.S.; FariS, L.P.; De Oliveira, R.N.; Pena, L. Ivermectin: An award-winning drug with expected antiviral activity against COVID-19. J. Control. Release 2021, 329, 758–761. [Google Scholar] [CrossRef]
- Hazan, S. Microbiome-based hypothesis on Ivermectin’s mechanism in COVID-19: Ivermectin feeds Bifidobacteria to boost immunity. Front. Microbiol. 2022, 13, 952321. [Google Scholar] [CrossRef]
- Zheng, M.; Schultz, M.B.; Sinclair, D.A. NAD+ in COVID-19 and viral infections. Trends Immunol. 2022, 43, 283–295. [Google Scholar] [CrossRef]
- Daoust, L.; Pilon, G.; Marette, A. Perspective: Nutritional strategies targeting the gut microbiome to mitigate COVID-19 outcomes. Adv. Nutr. 2021, 12, 1074–1086. [Google Scholar] [CrossRef]
- Gang, J.; Wang, H.; Xue, X.; Zhang, S. Microbiota and COVID-19: Long-term and complex influencing factors. Front. Microbiol. 2022, 13, 963488. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, L.; Wang, Y.; Dai, T.; Qin, Z.; Zhou, F.; Zhang, L. Alterations in microbiota of patients with COVID-19: Potential and therapeutic interventions. Signal Transduct. Target. Ther. 2022, 7, 143. [Google Scholar] [CrossRef]
- Maeda, Y.; Motooka, D.; Kawasaki, T.; Oki, H.; Noda, Y.; Adachi, Y.; Niitsu, T.; Okamoto, S.; Tanaka, K.; Fukushima, K.; et al. Longitudinal alterations of the gut mycobiota and microbiota on COVID-19 severity. BMC Infect. Dis. 2022, 22, 572. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, G.; Zhao, L.; Wang, W. Nutritional modulation of gut microbiota alleviates severe gastrointestinal symptoms in patients with post-acute COVID-19 syndrome. mBio 2022, 13, e0380121. [Google Scholar] [CrossRef]
- Haran, J.P.; Zheng, Y.; Knobil, K.; Palma, N.A.; Lawrence, J.F.; Wingertzahn, M.A. Targeting the microbiome with KB109 in outpatients with mild to moderate COVID-19 reduced medically attended acute care visits and improved symptoms in patients with comorbidities. medRxiv 2021. [Google Scholar] [CrossRef]
- Hashimoto, T.; Perlot, T.; Rehman, A.; Trichereau, J.; Ishiguro, H.; Paolino, M.; Sigl, V.; Hanada, T.; Hanada, R.; Lipinski, S.; et al. ACE2 links amino acid malnutrition to microbial ecology and intestinal inflammation. Nature 2012, 487, 477–481. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Lamers, M.M.; Beumer, J.; van der Vaart, J.; Knoops, K.; Puschhof, J.; Breugem, T.I.; Ravelli, R.B.G.; van Schayck, J.P.; Mykytyn, A.Z.; Duimel, H.Q.; et al. SARS-CoV-2 productively infects human gut enterocytes. Science 2020, 369, 50–54. [Google Scholar] [CrossRef]
- Penninger, J.M.; Grant, M.B.; Sung, J.J.Y. The role of angiotensin converting enzyme 2 in modulating gut microbiota, intestinal inflammation, and coronavirus infection. Gastroenterology 2021, 160, 39–46. [Google Scholar] [CrossRef]
- Foolchand, A.; Mazaleni, S.; Ghazi, T.; Chuturgoon, A.A. A review: Highlighting the links between epigenetics, COVID-19 infection, and vitamin D. Int. J. Mol. Sci. 2022, 23, 12292. [Google Scholar] [CrossRef] [PubMed]
- Karagiannis, F.; Peukert, K.; Surace, L.; Michla, M.; Nikolka, F.; Fox, M.; Weiss, P.; Feuerborn, C.; Maier, P.; Schulz, S.; et al. Impaired ketogenesis ties metabolism to T cell dysfunction in COVID-19. Nature 2022, 609, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Vargas, J.; Shapira, T.; Olmstead, A.D.; Villanueva, I.; Thompson, C.A.H.; Ennis, S.; Gao, G.; De Guzman, J.; Williams, D.E.; Wang, M. Discovery of lead natural products for developing pan-SARS-CoV-2 therapeutics. Antivir. Res. 2023, 209, 105484. [Google Scholar] [CrossRef]
- Blomberg, B.; Mohn, K.G.-I.; Brokstad, K.A.; Zhou, F.; Linchausen, D.W.; Hansen, B.-A.; Lartey, S.; Onyango, T.B.; Kuwelker, K.; Saeqvik, M.; et al. Long COVID in a prospective cohort of home-isolated patients. Nat. Med. 2021, 27, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, S.; Wu, Z.; Shang, Y.; Dong, X.; Li, G.; Zhang, L.; Chen, Y.; Ye, X.; Du, H.; et al. Clinical outcomes of COVID-19 in Wuhan, China: A large cohort study. Ann. Intensive Care 2020, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, Y.; Katano, H.; Zou, P.; Hohman, P.; Marques, A.; Tyring, S.K.; Follmann, D.; Cohen, J.I. Long-term administration of valacyclovir reduces the number of Epstein-Barr virus (EBV)-infected B cells but not the number of DNA copies per B cell in healthy volunteers. J. Virol. 2009, 83, 11857–11861. [Google Scholar] [CrossRef] [Green Version]
- Verna, D.; Thompson, J.; Swaminathan, S. Spironolactone blocks Epstein-Barr virus production by inhibiting EBV SM protein function. Proc. Natl. Acad. Sci. USA 2016, 113, 3609–3614. [Google Scholar] [CrossRef] [Green Version]
- Kottis, K.; Lechowicz, K.; Drozdzal, S.; Niedzwiedzka-Rystwej, P.; Wojdacz, T.K.; Grywalska, E.; Biernawska, J.; Wisniewska, M.; Parczewski, M. COVID-19: The potential beneficial therapeutic effects of Spironolactone during SARS-CoV-2 infection. Pharmaceuticals 2021, 14, 71. [Google Scholar]
- Mareev, V.Y.; Orlova, Y.A.; Plisyk, A.G.; Pavlikova, E.P.; Matskeplishvili, S.T.; Akopyan, Z.A.; Seredenina, E.M.; Potapenko, A.V.; Agapov, M.A.; Asratyan, D.A.; et al. Results of open-label non-randomized comparative clinical trial: BromhexIne and Spironolactone for CoronavirUs Infection requiring hospiTalization (BISCUIT). Kardologia 2020, 60, 4–15. [Google Scholar] [CrossRef]
- Dinnon, K.H.; Leist, S.R.; Okuda, K.; Dang, H.; Fritch, E.J.; Gully, K.L.; De la Cruz, G.; Evangelista, M.D.; Asakura, T.; Gilmore, R.C.; et al. SARS-CoV-2 infection produces chronic pulmonary epithelial and immune cell dysfunction with fibrosis in mice. Sci. Transl. Med. 2022, 14, eabo5070. [Google Scholar] [CrossRef]
- National Center for Advancing Translational Sciences (NCATS). NIH Begins Large Clinical Trial to Test Immune Modulators for Treatment of COVID-19; National Institutes of Health (NIH): Bethesda, MD, USA, 2020. Available online: https://www.nih.gov/news-events/news-releases/nih-begins-large-clinical-trial-test-immune-modulators-treatment-covid-19 (accessed on 19 September 2022).
- National Institutes of Health (NIH). Immune Modulator Drugs Improved Survival for People Hospitalized with COVID-19; National Institutes of Health (NIH): Bethesda, MD, USA, 2022. Available online: https://www.nih.gov/news-events/news-releases/immune-modulator-drugs-improved-survival-people-hospitalized-covid-19 (accessed on 19 September 2022).
- Pinto, M.D.; Lambert, N.; Downs, C.A.; Abrahim, H.; Hughes, T.D.; Rahmani, A.M.; Burton, C.W.; Chakraborty, R. Antihistamines for postacute sequelae of SARS-CoV-2 infection. J. Nurse Pract. 2022, 18, 335–338. [Google Scholar] [CrossRef] [PubMed]
- Glynne, P.; Tahmasebi, N.; Gant, V.; Gupta, R. Long COVID following mild SARS-CoV-2 infection: Characteristic T cell alterations and response to antihistamines. J. Investig. Med. 2021, 70, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Interim Guidance on Evaluating and Caring for Patients with Post-COVID Conditions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-care/post-covidindex.html (accessed on 11 October 2022).
- Herman, E.; Shih, E.; Cheng, A. Long COVID: Rapid evidence review. Am. Fam. Physician 2022, 106, 523–553. [Google Scholar] [PubMed]
- Herrera, J.E.; Niehaus, W.N.; Whiteson, J.; Azola, A.; Baratta, J.M.; Fleming, T.K.; Kim, S.Y.; Naqvi, H.; Sampsel, S.; Silver, J.K.; et al. Multidisciplinary collaborative consensus guidance statement on the assessment and treatment of fatigue in postacute sequelae of SARS-CoV-2 infection (PASC). Pm R 2021, 13, 1027–1043, corrected in Pm R 2022, 14, 164. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.R.; Sami, S.; Vuong, N.; Pathela, P.; Weiss, D.; Morgenthau, B.M.; Henseler, R.A.; Daskalakis, D.C.; Atas, J.; Patel, A.; et al. Lack of antibodies to severe acute respiratory syndrome coronavirus 2 (SARSCoV-2) in a large cohort of previously infected persons. Clin. Infect. Dis. 2021, 73, e3066–e3073. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, T.; Knight, M. Long COVID: A primer for family physicians. Am. Fam. Physician. 2020, 102, 716–717. [Google Scholar]
- Davis, H.E.; Assaf, G.S.; McCorkell, L.; Wei, H.; Low, R.J.; Re’em, Y.; Redfield, S.; Austin, J.P.; Akrami, A. Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EclinicalMedicine 2021, 38, 101019. [Google Scholar] [CrossRef]
- Vance, H.; Maslach, A.; Stoneman, E.; Harmes, K.; Ransom, A.; Seagly, K.; Furst, W. Addressing post-COVID symptoms: A guide for primary care physicians. J. Am. Board Fam. Med. 2021, 34, 1229–1242. [Google Scholar] [CrossRef]
- Fine, J.S.; Ambrose, A.F.; Didehbani, N.; Fleming, T.K.; Glashan, L.; Longo, M.; Merlino, A.; Ng, R.; Nora, G.J.; Rolin, S.; et al. Multi-disciplinary collaborative consensus guidance statement on the assessment and treatment of cognitive symptoms in patients with post-acute sequelae of SARS-CoV-2 infection (PASC). PM R. 2022, 14, 96–111. [Google Scholar] [CrossRef]
- Groff, D.; Sun, A.; Ssentongo, A.E.; Ba, D.M.; Parsons, N.; Poudel, G.R.; Lekoubou, A.; Oh, J.S.; Ericson, J.E.; Ssentongo, P.; et al. Short-term and long-term rates of postacute sequelae of SARS-CoV-2 infection: A systematic review. JAMA Netw. Open 2021, 4, e2128568. [Google Scholar] [CrossRef]
- Gaylis, N.B.; Ritter, A.; Kelly, S.A.; Pourhassan, N.Z.; Tiwary, M.; Sacha, J.B.; Hansen, S.G.; Recknor, C.; Yang, O.O. Reduced cell surface levels of C-C chemokine receptor 5 and immunosuppression in long coronavirus disease 2019 syndrome. Clin. Infect. Dis. 2022, 75, 1232–1234. [Google Scholar] [CrossRef] [PubMed]
- Gil, S.; Gualano, B.; Ladeira de Araujo, A.; Nascimento de Oliveira, G., Jr.; Damiano, R.F.; Pinna, F.; Imamura, M.; Rocha, V.; Kallas, E.; Batistella, L.R.; et al. Post-acute sequelae of SARS-CoV-2 associates with physical inactivity in a cohort of COVID-19 survivors. Sci. Rep. 2023, 13, 215. [Google Scholar] [CrossRef] [PubMed]
- Scurati, R.; Papini, N.; Alberti, G.; Tringali, C. The challenge of long COVID-19 management: From disease molecular hallmarks to the proposal of exercise as therapy. Int. J. Mol. Sci. 2022, 23, 12311. [Google Scholar] [CrossRef] [PubMed]
- Iddir, M.; Brito, A.; Dingeo, G.; Del Campo, S.S.F.; Samouda, H.; La Frano, M.R.; Bohn, T. Strengthening the immune system and reducing inflammation and oxidative stress through diet and nutrition: Considerations during the COVID-19 crisis. Nutrients 2020, 12, 1562. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Lyle, B.J.; Madsen, K.L.; Sonnenburg, J.; Verbeke, K.; Wu, G.D. Role for diet in normal gut barrier function: Developing guidance within the framework of food-labeling regulations. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G17–G39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Z.; Mak, J.W.Y.; Chan, F.K.L.; Ng, S.C. SIM01 as a novel microbiome replacement therapy for COVID-19: An open-label pilot study. J. Gastroenterol. Hepatol. 2021, 36, 272. [Google Scholar]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): A review. JAMA 2020, 323, 1824–1836. [Google Scholar] [CrossRef]
- Zoufaly, A.; Poglitsch, M.; Aberle, J.H.; Hoepler, W.; Seitz, T.; Traugott, M.; Grieb, A.; Pawelka, E.; Laferi, H.; Wenisch, C. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir. Med. 2020, 8, 1154–1158. [Google Scholar] [CrossRef]
- Verma, A.; Xu, K.; Du, T.; Zhu, P.; Liang, Z.; Liao, S.; Zhang, J.; Raizada, M.K.; Grant, M.B.; Li, Q. Expression of human ACE2 in Lactobacillus and beneficial effects in diabetic retinopathy in mice. Mol. Ther. Methods Clin. Dev. 2019, 14, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Bilinski, J.; Winter, K.; Jasinski, M.; Szczes, A.; Bilinska, N.; Mullish, B.H.; Malecka-Panas, E.; Basak, G.W. Rapid resolution of COVID-19 after faecal microbiota transplantation. Gut 2022, 71, 230–232. [Google Scholar] [CrossRef]
- Ianiro, G.; Mullish, B.H.; Kelly, C.R.; Kassam, Z.; Kujiper, E.J.; Ng, S.C.; Iqbal, T.H.; Allegretti, J.R.; Bibbó, S.; Sokol, H.; et al. Reorganisation of faecal microbiota transplant services during the COVID-19 pandemic. Gut 2020, 69, 1555–1563. [Google Scholar] [CrossRef]
- Laursen, R.P.; Hojsak, I. Probiotics for respiratory tract infections in children attending day care centers—A systematic review. Eur. J. Pediatr. 2018, 177, 979–994. [Google Scholar] [CrossRef]
- De Filippis, F.; Pellegrini, N.; Vannini, L.; Jeffery, I.B.; La Storia, A.; Laghi, L.; Serrazenetti, D.I.; Di Cagno, R.; Ferrocino, I.; Lazzi, C.; et al. High-level adherence to a Mediterranean diet beneficially impacts the gut microbiota and associated metabolome. Gut 2016, 65, 1812–1821. [Google Scholar] [CrossRef]
- Alvarado, D.M.; Son, J.; Thackray, L.B.; Diamond, M.S.; Ding, S.; Ciorba, M.A. IBD Investigators Group at Washington University. Mesalamine reduces intestinal ACE2 expression without modifying SARS-CoV-2 infection or disease severity in mice. bioRxiv 2021. bioRxiv:2021.07.23.453393. [Google Scholar]
- Kudryavtsev, I.; Rubinstein, A.; Golovkin, A.; Kalinina, O.; Vasilyev, K.; Rudenko, L.; Isakova-Sivak, I. Dysregulated immune responses in SARS-CoV-2-infected patients: A comprehensive overview. Viruses 2022, 14, 1082. [Google Scholar] [CrossRef]
- Malkova, A.; Kudryavtsev, I.; Starshinova, A.; Kudlay, D.; Zinchenko, Y.; Glushkova, A.; Yablonskiy, P.; Shoenfeld, Y. Post COVID-19 Syndrome in Patients with Asymptomatic/Mild Form. Pathogens 2021, 10, 1408. [Google Scholar] [CrossRef]
- Maglietta, G.; Diodati, F.; Puntoni, M.; Lazzarelli, S.; Marcomini, B.; Patrizi, L.; Caminiti, C. Prognostic factors for post-COVID-19 syndrome: A systematic review and meta-analysis. J. Clin. Med. 2022, 11, 1541. [Google Scholar] [CrossRef]
- Sandler, C.X.; Wyller, V.B.B.; Moss-Morris, R.; Buchwald, D.; Crawley, E.; Hautvast, J.; Katz, B.Z.; Knoop, H.; Little, P.; Taylor, R.; et al. Long COVID and post-infective fatigue syndrome: A review. Open Forum Infect. Dis. 2021, 8, ofab440. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Nearly One in Five American Adults Who Have Had COVID-19 Still Have “Long COVID”. Available online: https://www.cdc.gov/nchs/pressroom/nchs_press_releases/2022/20220622.htm (accessed on 4 October 2022).
- Datta, S.D.; Talwar, A.; Lee, J.T. A proposed framework and timeline of the spectrum of disease due to SARS-CoV-2 infection: Illness beyond acute infection and public health implications. JAMA 2020, 324, 2251–2252. [Google Scholar] [CrossRef]
- Darby, J.B.; Jackson, J.M. Kawasaki disease and multisystem inflammatory syndrome in children: An overview and comparison. Am. Fam. Physician. 2021, 104, 244–252. [Google Scholar]
- Payne, A.B.; Gilani, Z.; Godfred-Cato, S.; Belay, E.D.; Feldstein, L.R.; Patel, M.M.; Randolph, A.G.; Newhams, M.; Thomas, D.; Magleby, R.; et al. Incidence of multisystem inflammatory syndrome in children among US persons infected with SARS-CoV-2. JAMA Netw. Open 2021, 4, e2116420. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. Infectious Diseases Society of America. Multisystem Inflammatory Syndrome in Adults (MIS-A). Updated June 24, 2022. Available online: https://www.idsociety.org/covid-19-real-time-learning-network/disease-manifestations--complications/multisystem-inflammatory-syndrome-inadults-mis-a/ (accessed on 31 August 2022).
- Nath, A. Long-haul COVID. Neurology 2020, 95, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Tenforde, M.W.; Kim, S.S.; Lindsell, C.J.; Rose, E.B.; Shapiro, N.I.; Files, D.C.; Gibbs, K.W.; Erickson, H.L.; Steingrub, J.S.; Smithline, H.A.; et al. IVY Network Investigators; CDC COVID-19 Response Team. Symptom duration and risk factors for delayed return to usual health among outpatients with COVID-19 in a multistate health care systems network—United States, March-June 2020. MMWR Morb. Mortal Wkly. Rep. 2020, 69, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Aiyegbusi, O.L.; Hughes, S.E.; Turner, G.; Rivera, S.C.; McMullan, C.; Chandan, J.S.; Haroon, S.; Price, G.; Davies, E.H.; Nirantharakumar, K.; et al. Symptoms, complications and management of long COVID: A review. J. R. Soc. Med. 2021, 114, 428–442. [Google Scholar] [CrossRef]
- Antonelli, M.; Penfold, R.S.; Merino, J.; Sudre, C.H.; Molteni, E.; Bewrry, S.; Canas, L.S.; Graham, M.S.; Klaser, K.; Modat, M.; et al. Risk factors and disease profile of post-vaccination SARS-CoV-2 infection in UK users of the COVID Symptom Study app: A prospective, community-based, nested, case-control study. Lancet Infect. Dis. 2022, 22, 43–55. [Google Scholar] [CrossRef]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long COVID-mechanisms, risk factors, and management. BMJ 2021, 374, n1648, corrected in BMJ 2021, 374, n1944. [Google Scholar] [CrossRef]
- Kamal, M.; Omirah, M.A.; Hussein, A.; Saeed, H. Assessment and characterisation of post-COVID-19 manifestations. Int. J. Clin. Pract. 2021, 75, e13746. [Google Scholar] [CrossRef]
- Sneller, M.C.; Liang, C.J.; Marques, A.R.; Chung, J.Y.; Shanbhag, S.M.; Fontsana, J.R.; Raza, H.; Okeke, O.; Dewar, R.L.; Higgins, B.P.; et al. A longitudinal study of COVID-19 sequelae and immunity: Baseline findings. Ann. Intern. Med. 2022, 175, 969–979. [Google Scholar] [CrossRef]
- Albrich, W.C.; Ghosh, T.S.; Ahearn-Ford, S.; Mikaeloff, F.; Lunjani, N.; Forde, B.; Suh, N.; Kleger, G.-R.; Pietsch, U.; Frischknecht, M.; et al. A high-risk gut microbiota configuration associates with fatal hyperinflammatory immune and metabolic responses to SARS-CoV-2. Gut Microbes 2022, 14, e2073131. [Google Scholar] [CrossRef]
- Lau, H.C.-H.; Ng, S.C.; Yu, J. Targeting the gut microbiota in coronavirus disease 2019: Hype or hope? Gastroenterology 2022, 162, 9–16. [Google Scholar] [CrossRef]
- Koutsakos, M.; Rowntree, L.C.; Hensen, L.; Chua, B.Y.; van de Sandt, C.E.; Habel, J.R.; Zhang, W.; Jia, X.; Kedzierski, L.; Ashhurst, T.M.; et al. Integrated immune dynamics define correlates of COVID-19 severity and antibody responses. Cell. Rep. Med. 2021, 2, 100208. [Google Scholar] [CrossRef]
- Leviatan, S.; Segai, E. Identifying gut microbes that affect human health. Nature 2020, 587, 373–374. [Google Scholar] [CrossRef]
- Altmae, S.; Franasiak, J.M.; Mandar, R. The seminal microbe in health and disease. Nat. Rev. Urol. 2019, 16, 703–721. [Google Scholar] [CrossRef]
- Van Treuren, W.; Dodd, D. Microbial contribution to the human metabolome: Implications for health and disease. Annu. Rev. Pathol. 2020, 15, 345–369. [Google Scholar] [CrossRef] [Green Version]
- Nie, P.; Li, Z.; Wang, Y.; Zhang, Y.; Zhao, M.; Luo, J.; Du, S.; Deng, Z.; Chen, J.; Wang, Y.; et al. Gut microbiome interventions in human health and diseases. Med. Res. Rev. 2019, 39, 2286–2313. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell biology of tight junction barrier regulation and mucosal disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef]
- Velikova, T.; Snegarova, V.; Kukov, A.; Batselova, H.; Mihova, A.; Nakov, R. Gastrointestinal mucosal immunity and COVID-19. World J. Gastroenterol. 2021, 27, 5047–5059. [Google Scholar] [CrossRef]
- Elizalde-Diaz, J.P.; Miranda-Narvaez, C.L.; Martinez-Lazcano, J.C.; Martinez-Martinez, E. The relationship between chronic immune response and neurodegenerative damage in long COVID-19. Front. Immunol. 2022, 13, 1039427. [Google Scholar] [CrossRef]
- Zhao, B.; Zhong, M.; Yang, Q.; Hong, K.; Xia, J.; Li, X.; Liu, Y.; Chen, Y.Q.; Yang, J.; Huang, C.; et al. Alterations in Phenotypes and Responses of T Cells Within 6 Months of Recovery from COVID-19: A Cohort Study. Virol. Sin. 2021, 36, 859–868. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Shared Peptide | SARS-CoV-2 Protein (UniProt ID) | Human Chaperone (UniProt ID) | Putative Epitope |
|---|---|---|---|
| EIPKEE | Replicase polyprotein 1ab [P0DTD1] | Heat shock 60 kDa protein [P10809] | AEIPKEEVKPFITESKPSVEQRKQDDKK |
| LPYPDP | Replicase polyprotein 1ab [P0DTD1] | Heat shock 70 kDa protein [P34932] | TLEAYYSSPQDLPYPDPAIA |
| KDKKKK | Nucleocapsid phospho-protein [P0DTD1] | Heat shock protein HSP 90-beta [P08238] | KTFPPTEPKDKKKKADETQALPQRQKKQQ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vojdani, A.; Vojdani, E.; Saidara, E.; Maes, M. Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID. Viruses 2023, 15, 400. https://doi.org/10.3390/v15020400
Vojdani A, Vojdani E, Saidara E, Maes M. Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID. Viruses. 2023; 15(2):400. https://doi.org/10.3390/v15020400
Chicago/Turabian StyleVojdani, Aristo, Elroy Vojdani, Evan Saidara, and Michael Maes. 2023. "Persistent SARS-CoV-2 Infection, EBV, HHV-6 and Other Factors May Contribute to Inflammation and Autoimmunity in Long COVID" Viruses 15, no. 2: 400. https://doi.org/10.3390/v15020400