
Covalent Inhibitors from Saudi Medicinal Plants Target RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2
 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Details of Phytochemcials and Prepartion of Ligands
2.2. Preparation of RdRP
2.3. Docking Experiments
2.3.1. Docking Validation
2.3.2. Non-Covalent Docking
2.3.3. Irreversible (Covalent) Docking
2.4. MD Simulation Studies
3. Results
3.1. Details of Phytochemicals (Compounds 1–10)
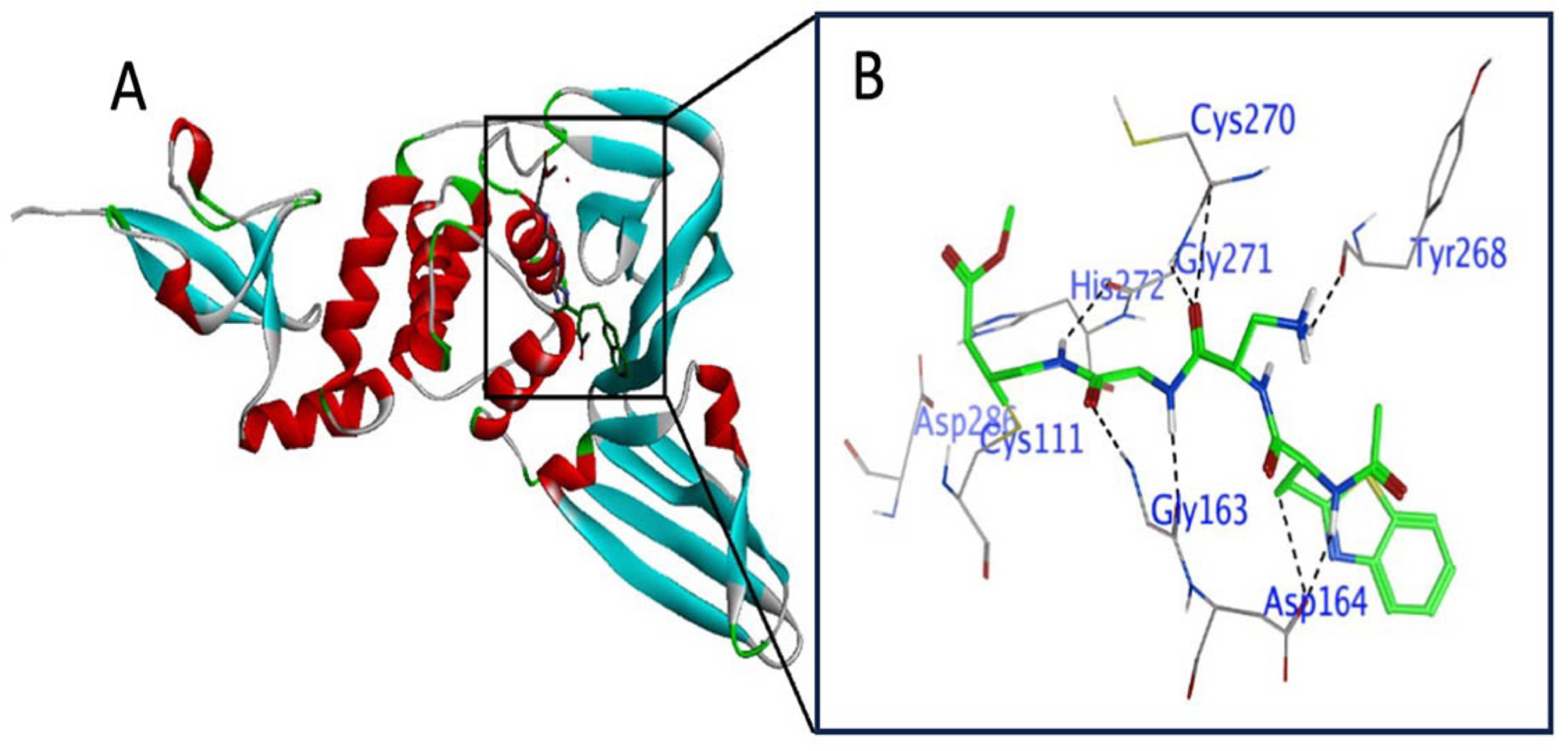
3.2. Active Site Explanation in RdRP
3.3. Docking Protocol Validation with RdRP
3.4. Rreversible (Non-Covalent) Docking of Compounds 1–10 with RdRP
3.5. Irreversible (Covalent) Docking of Compounds 1–10 with RdRP
3.6. Stability, Conformity, and Compactness Analysis by MD Simulation
3.6.1. RMSD Measurements
3.6.2. Quantification of RMSF and Rg
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Bi, Q.; Wu, Y.; Mei, S.; Ye, C.; Zou, X.; Zhang, Z.; Liu, X.; Wei, L.; Truelove, S.A.; Zhang, T.; et al. Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: A retrospective cohort study. Lancet Infect. Dis. 2020, 20, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Lu, H. Drug treatment options for the 2019-new coronavirus (2019-nCoV). Biosci. Trends 2020, 14, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.-J.; Li, P.; Song, J.; Zhang, S.-Y.; Xie, H.-Z. Mechanisms of synergistic neurotoxicity induced by two high risk pesticide residues—Chlorpyrifos and Carbofuran via oxidative stress. Toxicol. Vitr. 2019, 54, 338–344. [Google Scholar] [CrossRef]
- WHO. WHO Coronavirus (COVID-19) Dashboard. 2023. Available online: https://covid19.who.int (accessed on 9 October 2023).
- Monteleone, G.; Sarzi-Puttini, P.C.; Ardizzone, S. Preventing COVID-19-induced pneumonia with anticytokine therapy. Lancet Rheumatol. 2020, 2, e255–e256. [Google Scholar] [CrossRef]
- Amaral-Machado, L.; Oliveira, W.N.; Rodrigues, V.M.; Albuquerque, N.A.; Alencar, É.N.; Egito, E.S.T. Could natural products modulate early inflammatory responses, preventing acute respiratory distress syndrome in COVID-19-confirmed patients? Biomed. Pharmacother. 2021, 134, 111143. [Google Scholar] [CrossRef]
- Tang, T.; Bidon, M.; Jaimes, J.A.; Whittaker, G.R.; Daniel, S. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antivir. Res. 2020, 178, 104792. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Veesler, D. Chapter Four—Structural insights into coronavirus entry. In Advances in Virus Research; Rey, F.A., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 105, pp. 93–116. [Google Scholar]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020, 26, 681–687. [Google Scholar] [CrossRef]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef]
- Das, G.; Das, T.; Chowdhury, N.; Chatterjee, D.; Bagchi, A.; Ghosh, Z. Repurposed drugs and nutraceuticals targeting envelope protein: A possible therapeutic strategy against COVID-19. Genomics 2021, 113, 1129–1140. [Google Scholar] [CrossRef] [PubMed]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar] [CrossRef]
- Jade, D.; Ayyamperumal, S.; Tallapaneni, V.; Joghee Nanjan, C.M.; Barge, S.; Mohan, S.; Nanjan, M.J. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur. J. Pharmacol. 2021, 901, 174082. [Google Scholar] [CrossRef] [PubMed]
- Tumskiy, R.S.; Tumskaia, A.V.; Klochkova, I.N.; Richardson, R.J. SARS-CoV-2 proteases Mpro and PLpro: Design of inhibitors with predicted high potency and low mammalian toxicity using artificial neural networks, ligand-protein docking, molecular dynamics simulations, and ADMET calculations. Comput. Biol. Med. 2023, 153, 106449. [Google Scholar] [CrossRef]
- Lu, J.; Lu, W.; Jiang, H.; Yang, C.; Dong, X. Molecular Docking and Dynamics of Phytochemicals From Chinese Herbs with SARS-CoV-2 RdRp. Nat. Prod. Commun. 2022, 17, 1934578X221105693. [Google Scholar] [CrossRef]
- Nesterenko, P.A.; McLaughlin, J.; Tsai, B.L.; Burton Sojo, G.; Cheng, D.; Zhao, D.; Mao, Z.; Bangayan, N.J.; Obusan, M.B.; Su, Y.; et al. HLA-A∗02:01 restricted T cell receptors against the highly conserved SARS-CoV-2 polymerase cross-react with human coronaviruses. Cell Rep. 2021, 37, 110167. [Google Scholar] [CrossRef]
- Zhang, L.-C.; Zhao, H.-L.; Liu, J.; He, L.; Yu, R.-L.; Kang, C.-M. Design of SARS-CoV-2 Mpro, PLpro dual-target inhibitors based on deep reinforcement learning and virtual screening. Future Med. Chem. 2022, 14, 393–405. [Google Scholar] [CrossRef]
- Glab-ampai, K.; Kaewchim, K.; Thavorasak, T.; Saenlom, T.; Thepsawat, W.; Mahasongkram, K.; Thueng-In, K.; Sookrung, N.; Chaicumpa, W.; Chulanetra, M. Targeting Emerging RNA Viruses by Engineered Human Superantibody to Hepatitis C Virus RNA-Dependent RNA Polymerase. Front. Microbiol. 2022, 13, 926929. [Google Scholar] [CrossRef]
- Aziz, S.; Waqas, M.; Mohanta, T.K.; Halim, S.A.; Iqbal, A.; Ali, A.; Khalid, A.; Abdalla, A.N.; Khan, A.; Al-Harrasi, A. Identifying non-nucleoside inhibitors of RNA-dependent RNA-polymerase of SARS-CoV-2 through per-residue energy decomposition-based pharmacophore modeling, molecular docking, and molecular dynamics simulation. J. Infect. Public Health 2023, 16, 501–519. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Kuno-Maekawa, M.; Sangawa, H.; Uehara, S.; Kozaki, K.; Nomura, N.; Egawa, H.; Shiraki, K. Mechanism of Action of T-705 against Influenza Virus. Antimicrob. Agents Chemother. 2005, 49, 981–986. [Google Scholar] [CrossRef]
- Furuta, Y.; Komeno, T.; Nakamura, T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B 2017, 93, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef]
- De Clercq, E.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Clark, M.P.; Ledeboer, M.W.; Davies, I.; Byrn, R.A.; Jones, S.M.; Perola, E.; Tsai, A.; Jacobs, M.; Nti-Addae, K.; Bandarage, U.K.; et al. Discovery of a Novel, First-in-Class, Orally Bioavailable Azaindole Inhibitor (VX-787) of Influenza PB2. J. Med. Chem. 2014, 57, 6668–6678. [Google Scholar] [CrossRef] [PubMed]
- Lemm Julie, A.; Liu, M.; Gentles Robert, G.; Ding, M.; Voss, S.; Pelosi Lenore, A.; Wang, Y.-K.; Rigat Karen, L.; Mosure Kathleen, W.; Bender John, A.; et al. Preclinical Characterization of BMS-791325, an Allosteric Inhibitor of Hepatitis C Virus NS5B Polymerase. Antimicrob. Agents Chemother. 2014, 58, 3485–3495. [Google Scholar] [CrossRef] [PubMed]
- Cerón-Carrasco, J.P. When Virtual Screening Yields Inactive Drugs: Dealing with False Theoretical Friends. ChemMedChem 2022, 17, e202200278. [Google Scholar] [CrossRef]
- Kumar, S.; Kashyap, P.; Chowdhury, S.; Kumar, S.; Panwar, A.; Kumar, A. Identification of phytochemicals as potential therapeutic agents that binds to Nsp15 protein target of coronavirus (SARS-CoV-2) that are capable of inhibiting virus replication. Phytomedicine 2020, 85, 153317. [Google Scholar] [CrossRef]
- Mani, J.S.; Johnson, J.B.; Steel, J.C.; Broszczak, D.A.; Neilsen, P.M.; Walsh, K.B.; Naiker, M. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res. 2020, 284, 197989. [Google Scholar] [CrossRef]
- Ho, T.Y.; Wu, S.L.; Chen, J.C.; Li, C.C.; Hsiang, C.Y. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antivir. Res. 2007, 74, 92–101. [Google Scholar] [CrossRef]
- Gowrishankar, S.; Muthumanickam, S.; Kamaladevi, A.; Karthika, C.; Jothi, R.; Boomi, P.; Maniazhagu, D.; Pandian, S.K. Promising phytochemicals of traditional Indian herbal steam inhalation therapy to combat COVID-19—An in silico study. Food Chem. Toxicol. 2021, 148, 111966. [Google Scholar] [CrossRef] [PubMed]
- Panikar, S.; Shoba, G.; Arun, M.; Sahayarayan, J.J.; Nanthini, A.U.R.; Chinnathambi, A.; Alharbi, S.A.; Nasif, O.; Kim, H.-J. Essential oils as an effective alternative for the treatment of COVID-19: Molecular interaction analysis of protease (Mpro) with pharmacokinetics and toxicological properties. J. Infect. Public Health 2021. [Google Scholar] [CrossRef] [PubMed]
- Parida, P.K.; Paul, D.; Chakravorty, D. Nature’s therapy for COVID-19: Targeting the vital non-structural proteins (NSP) from SARS-CoV-2 with phytochemicals from Indian medicinal plants. Phytomed. Plus 2021, 1, 100002. [Google Scholar] [CrossRef]
- Azeem, M.; Mustafa, G.; Mahrosh, H.S. Virtual screening of phytochemicals by targeting multiple proteins of severe acute respiratory syndrome coronavirus 2: Molecular docking and molecular dynamics simulation studies. Int. J. Immunopathol. Pharmacol. 2022, 36, 3946320221142793. [Google Scholar] [CrossRef]
- Chen, T.-H.; Tsai, M.-J.; Chang, C.-S.; Xu, L.; Fu, Y.-S.; Weng, C.-F. The exploration of phytocompounds theoretically combats SARS-CoV-2 pandemic against virus entry, viral replication and immune evasion. J. Infect. Public Health 2023, 16, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Rajak, P.; Ganguly, A. In silico study unfolds inhibitory potential of epicatechin gallate against SARS-CoV-2 entry and replication within the host cell. Mechanobiol. Med. 2023, 1, 100015. [Google Scholar] [CrossRef]
- Ahmed, S.; Al-Rehaily, A.J.; Ahmad, M.S.; Yousaf, M.; Nur-e-Alam, M.; Parvez, M.K.; Al-Dosari, M.S.; Noman, O.M.; Khan, S.I.; Khan, I.A. Chemical constituents from Oncocalyx glabratus and their biological activities. Phytochem. Lett. 2017, 20, 128–132. [Google Scholar] [CrossRef]
- Ahmed, S.; Al-Rehaily, A.J.; Ahmad, M.S.; Yousaf, M.; Nur-e-Alam, M.; Thomas, J.; Khan, S.I.; Khan, I.A. Cytotoxic and antiinflammatory activities of the chemical constituents isolated from Baccharoides schimperi DC. S. Afr. J. Bot. 2018, 114, 9–13. [Google Scholar] [CrossRef]
- Ahmed, S.; Mothana, R.; Yousaf, M.; Al-Rehaily, A. Activity guided isolation of chemical constituents from the biologically active methanol extract of Euphorbia schimperi c. presl. Bull. Chem. Soc. Ethiop. 2017, 31, 471–479. [Google Scholar] [CrossRef]
- ChemAxon. Available online: https://chemaxon.com/marvin (accessed on 3 September 2023).
- Mesecar, A.D. Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77. Protein Data Bank 2020. [Google Scholar] [CrossRef]
- Khalaf, H.S.; Naglah, A.M.; Al-Omar, M.A.; Moustafa, G.O.; Awad, H.M.; Bakheit, A.H. Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide. Molecules 2020, 25, 3589. [Google Scholar] [CrossRef]
- Al-Khodairy, F.M.; Khan, M.K.A.; Kunhi, M.; Pulicat, M.S.; Akhtar, S.; Arif, J.M. In Silico prediction of mechanism of Erysolin-induced apoptosis in human breast cancer cell lines. Am. J. Bioinform. Res. 2013, 3, 62–71. [Google Scholar]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins Struct. Funct. Bioinform. 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Wan, X.; Burlingame, A.L.; Taunton, J. Ligand Conformational Bias Drives Enantioselective Modification of a Surface-Exposed Lysine on Hsp90. J. Am. Chem. Soc. 2020, 142, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.; Lesyng, B. Generalized Born Model: Analysis, Refinement, and Applications to Proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein−Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- CHARMM General Force Field (CGenFF). Available online: https://cgenff.silcsbio.com (accessed on 3 September 2023).
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, S.; Onck, P.R.; Giessen, E.v.d. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Ghazwani, M.Y.; Bakheit, A.H.; Hakami, A.R.; Alkahtani, H.M.; Almehizia, A.A. Virtual Screening and Molecular Docking Studies for Discovery of Potential RNA-Dependent RNA Polymerase Inhibitors. Crystals 2021, 11, 471. [Google Scholar] [CrossRef]
- Naydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K.; et al. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef] [PubMed]
- Shitrit, A.; Zaidman, D.; Kalid, O.; Bloch, I.; Doron, D.; Yarnizky, T.; Buch, I.; Segev, I.; Ben-Zeev, E.; Segev, E.; et al. Conserved interactions required for inhibition of the main protease of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Sci. Rep. 2020, 10, 20808. [Google Scholar] [CrossRef]
- Hassam, M.; Bashir, M.A.; Shafi, S.; Zahra, N.-u.-A.; Khan, K.; Jalal, K.; Siddiqui, H.; Uddin, R. Identification of potent compounds against SARs-CoV-2: An in-silico based drug searching against Mpro. Comput. Biol. Med. 2022, 151, 106284. [Google Scholar] [CrossRef]
- Dampalla, C.S.; Miller, M.J.; Kim, Y.; Zabiegala, A.; Nguyen, H.N.; Madden, T.K.; Thurman, H.A.; Machen, A.J.; Cooper, A.; Liu, L.; et al. Structure-guided design of direct-acting antivirals that exploit the gem-dimethyl effect and potently inhibit 3CL proteases of severe acute respiratory syndrome Coronavirus-2 (SARS-CoV-2) and middle east respiratory syndrome coronavirus (MERS-CoV). Eur. J. Med. Chem. 2023, 254, 115376. [Google Scholar] [CrossRef]
- Brindani, N.; Munafò, F.; Menichetti, A.; Donati, E.; Nigro, M.; Ottonello, G.; Armirotti, A.; De Vivo, M. Design, synthesis, docking, and biochemical characterization of non-nucleoside SARS-CoV-2 RdRp inhibitors. Biorg. Med. Chem. 2023, 80, 117179. [Google Scholar] [CrossRef]
- WHO. Tracking SARS-CoV-2 Variants. 2023. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 9 October 2023).
- Taylor, P.C.; Adams, A.C.; Hufford, M.M.; de la Torre, I.; Winthrop, K.; Gottlieb, R.L. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol. 2021, 21, 382–393. [Google Scholar] [CrossRef]
- US FDA. COVID-19 Vaccines. 2019. Available online: https://www.fda.gov/emergency-preparedness-and-response/coronavirus-disease-2019-covid-19/covid-19-vaccines (accessed on 9 October 2023).
- Byléhn, F.; Menéndez, C.A.; Perez-Lemus, G.R.; Alvarado, W.; de Pablo, J.J. Modeling the Binding Mechanism of Remdesivir, Favilavir, and Ribavirin to SARS-CoV-2 RNA-Dependent RNA Polymerase. ACS Cent. Sci. 2021, 7, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Pang, Z.; Li, M.; Lou, F.; An, X.; Zhu, S.; Song, L.; Tong, Y.; Fan, H.; Fan, J. Molnupiravir and Its Antiviral Activity Against COVID-19. Front. Immunol. 2022, 13, 855496. [Google Scholar] [CrossRef]
- Duveau, D.Y.; Thomas, C.J. The Remarkable Selectivity of Nirmatrelvir. ACS Pharmacol. Transl. Sci. 2022, 5, 445–447. [Google Scholar] [CrossRef] [PubMed]
- McLean, G.; Kamil, J.; Lee, B.; Moore, P.; Schulz Thomas, F.; Muik, A.; Sahin, U.; Türeci, Ö.; Pather, S. The Impact of Evolving SARS-CoV-2 Mutations and Variants on COVID-19 Vaccines. mBio 2022, 13, e02979-21. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 2021, 184, 3086–3108. [Google Scholar] [CrossRef]
- ASPR, Administration for Strategic Preparedness & Response, FDA Approves First Oral Antiviral for Treatment of COVID-19 in Adults. 2023. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-oral-antiviral-treatment-covid-19-adults (accessed on 9 October 2023).
- Lee, T.C.; Morris, A.M.; Grover, S.A.; Murthy, S.; McDonald, E.G. Outpatient Therapies for COVID-19: How Do We Choose? Open Forum Infect. Dis. 2022, 9, ofac008. [Google Scholar] [CrossRef]
- ASPR, Administration for Strategic Preparedness & Response, Side-by-Side Overview of Therapeutics Authorized or Approved for the Treatment of Mild to Moderate COVID-19. 2023. Available online: https://aspr.hhs.gov/COVID-19/Therapeutics/Documents/side-by-side-overview.pdf (accessed on 9 October 2023).
- Zhang, Y.-N.; Zhu, G.-H.; Liu, W.; Xiong, Y.; Hu, Q.; Zhuang, X.-Y.; Jia, G.-H.; Zhang, W.-D.; Ge, G.-B. Discovery and characterization of the covalent SARS-CoV-2 3CLpro inhibitors from Ginkgo biloba extract via integrating chemoproteomic and biochemical approaches. Phytomedicine 2023, 114, 154796. [Google Scholar] [CrossRef]
- Barhouchi, B.; Menacer, R.; Bouchkioua, S.; Mansour, A.; Belattar, N. Compounds from myrtle flowers as antibacterial agents and SARS-CoV-2 inhibitors: In-vitro and molecular docking studies. Arab. J. Plant. Prot. 2023, 16, 104939. [Google Scholar] [CrossRef]
- Hossain, A.; Rahman, M.E.; Rahman, M.S.; Nasirujjaman, K.; Matin, M.N.; Faruqe, M.O.; Rabbee, M.F. Identification of medicinal plant-based phytochemicals as a potential inhibitor for SARS-CoV-2 main protease (Mpro) using molecular docking and deep learning methods. Comput. Biol. Med. 2023, 157, 106785. [Google Scholar] [CrossRef]
- Parihar, A.; Sonia, Z.F.; Akter, F.; Ali, M.A.; Hakim, F.T.; Hossain, M.S. Phytochemicals-based targeting RdRp and main protease of SARS-CoV-2 using docking and steered molecular dynamic simulation: A promising therapeutic approach for Tackling COVID-19. Comput. Biol. Med. 2022, 145, 105468. [Google Scholar] [CrossRef]
- Guimarães Santana, B.C.; de Almeida Marques, D.P.; dos Santos Freitas, A.; Ferreira, M.M.; de Sousa Lopes, D.; Bagno, F.F.; Guimarães da Fonseca, F.; dos Reis, J.G.A.C.; Oliveira Mendes, T.A.d.; Santos, J.L.d.; et al. Protease inhibitors from Theobroma cacao impair SARS-CoV-2 replication in vitro. Heliyon 2023, 9, e15860. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, J.; Ikram, S.; Ahmad, F.; Rehman, I.U.; Mushtaq, M. SARS-CoV-2 RNA Dependent RNA polymerase (RdRp)—A drug repurposing study. Heliyon 2020, 6, e04502. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Song, L.; Xu, H.; Fikatas, A.; Oeyen, M.; De Jonghe, S.; Zhao, F.; Jing, L.; Jochmans, D.; Vangeel, L.; et al. Identification of Polyphenol Derivatives as Novel SARS-CoV-2 and DENV Non-Nucleoside RdRp Inhibitors. Molecules 2023, 28, 160. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Padhi, A.K.; Shukla, R.; Saudagar, P.; Tripathi, T. High-throughput rational design of the remdesivir binding site in the RdRp of SARS-CoV-2: Implications for potential resistance. iScience 2021, 24, 101992. [Google Scholar] [CrossRef]
- Gado, S.; ALAGÖZ, Z. RNA-DEPENDENT RNA POLYMERASE (RDRP) INHIBITOR DRUGS AGAINST SARS-COV-2: A MOLECULAR DOCKING STUDY SARS-COV-2’YE KARŞI RNA-BAĞIMLI RNA POLİMERAZ (RDRP) İNHİBİTÖR İLAÇLARI: BİR MOLEKÜLER DOCKİNG ÇALIŞMASI. Ank. Univ. Eczaci. Fak. Derg. 2022, 46, 62–77. [Google Scholar] [CrossRef]
- Gordon, C.J.; Tchesnokov, E.P.; Woolner, E.; Perry, J.K.; Feng, J.Y.; Porter, D.P.; Götte, M. Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 2020, 295, 6785–6797. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Natural plant products as potential inhibitors of RNA dependent RNA polymerase of Severe Acute Respiratory Syndrome Coronavirus-2. PLoS ONE 2021, 16, e0251801. [Google Scholar] [CrossRef]
- Boadu, A.; Agoni, C.; Karpoormath, R.; Soliman, M.; Nlooto, M. Repurposing antiviral phytochemicals from the leaf extracts of Spondias mombin (Linn) towards the identification of potential SARSCOV-2 inhibitors. Sci. Rep. 2022, 12, 10896. [Google Scholar] [CrossRef]
- Scarpino, A.; Ferenczy, G.G.; Keserű, G.M. Comparative Evaluation of Covalent Docking Tools. J. Chem. Inf. Model. 2018, 58, 1441–1458. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, D.; Cheng, X. Recent Advances in Covalent Drug Discovery. Pharmaceuticals 2023, 16, 663. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. WIREs RNA 2013, 4, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Uma Reddy, B.; Routhu, N.K.; Kumar, A. Multifaceted roles of plant derived small molecule inhibitors on replication cycle of SARS-CoV-2. Microb. Pathog. 2022, 168, 105512. [Google Scholar] [CrossRef]
- Strelow, J.M. A Perspective on the Kinetics of Covalent and Irreversible Inhibition. SLAS Discov. 2017, 22, 3–20. [Google Scholar] [CrossRef]
- La Monica, G.; Bono, A.; Lauria, A.; Martorana, A. Targeting SARS-CoV-2 Main Protease for Treatment of COVID-19: Covalent Inhibitors Structure–Activity Relationship Insights and Evolution Perspectives. J. Med. Chem. 2022, 65, 12500–12534. [Google Scholar] [CrossRef]
- Shindo, N.; Ojida, A. Recent progress in covalent warheads for in vivo targeting of endogenous proteins. Biorg. Med. Chem. 2021, 47, 116386. [Google Scholar] [CrossRef]
- Kovacic, F.; Mandrysch, A.; Poojari, C.; Strodel, B.; Jaeger, K.-E. Structural features determining thermal adaptation of esterases. Protein Eng. Des. Sel. 2016, 29, 65–76. [Google Scholar] [CrossRef]
- Dong, Y.-W.; Liao, M.-l.; Meng, X.-L.; Somero, G.N. Structural flexibility and protein adaptation to temperature: Molecular dynamics analysis of malate dehydrogenases of marine molluscs. Proc. Natl. Acad. Sci. USA 2018, 115, 1274–1279. [Google Scholar] [CrossRef]
- Aouidate, A.; Ghaleb, A.; Chtita, S.; Aarjane, M.; Ousaa, A.; Maghat, H.; Sbai, A.; Choukrad, M.b.; Bouachrine, M.; Lakhlifi, T. Identification of a novel dual-target scaffold for 3CLpro and RdRp proteins of SARS-CoV-2 using 3D-similarity search, molecular docking, molecular dynamics and ADMET evaluation. J. Biomol. Struct. Dyn. 2021, 39, 4522–4535. [Google Scholar] [CrossRef]
- Mathpal, S.; Joshi, T.; Sharma, P.; Joshi, T.; Pundir, H.; Pande, V.; Chandra, S. A dynamic simulation study of FDA drug from zinc database against COVID-19 main protease receptor. J. Biomol. Struct. Dyn. 2022, 40, 1084–1100. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand (Compound 7) | Receptor (RdRp) | Interaction Type | Distance (Å) | Energy (kcal/mol) | Docking Score (kcal/mol) |
|---|---|---|---|---|---|
| O 32 | SD MET 601 (A) | H–donor | 3.47 | −0.5 | −6.136 |
| C 34 | O VAL 588 (A) | H–donor | 3.16 | −0.3 | |
| O 20 | CE LYS 593 (A) | H–acceptor | 3.27 | −0.3 | |
| C 5 | Six-ring PHE 812 (A) | H–π | 4.58 | −0.4 |
| Ligand (Compound 8) | Receptor (RdRp) | Interaction Type | Distance (Å) | Energy (kcal/mol) | Docking Score (kcal/mol) |
|---|---|---|---|---|---|
| C 51 | OD2 ASP 760 (A) | H–donor | 3.59 | −0.3 | −13.482 |
| O 57 | OD2 ASP 761 (A) | H–donor | 3.32 | −1.7 | |
| C 59 | OP2 G 5 (G) | H–donor | 3.28 | −0.6 | |
| C 15 | Five-ring G 3 (G) | H–π | 3.63 | −0.3 | |
| C 27 | Five-ring G 3 (G) | H–π | 4.37 | −0.3 |
| Ligand (Compound 9) | Receptor (RdRp) | Interaction Type | Distance (Å) | Energy (kcal/mol) | Docking Score (kcal/mol) |
|---|---|---|---|---|---|
| O 15 | OD2 ASP 761 (A) | H–donor | 3 | −1.1 | −6.732 |
| C 29 | O2 U −4 (F) | H–donor | 3.12 | −0.3 | |
| C 18 | Six-ring G 3 (G) | H–π | 4.42 | −0.3 | |
| C 19 | Five-ring G 5 (G) | H–π | 3.68 | −0.3 | |
| C 27 | Five-ring G 3 (G) | H–π | 4.35 | −0.5 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bakheit, A.H.; Saquib, Q.; Ahmed, S.; Ansari, S.M.; Al-Salem, A.M.; Al-Khedhairy, A.A. Covalent Inhibitors from Saudi Medicinal Plants Target RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2. Viruses 2023, 15, 2175. https://doi.org/10.3390/v15112175
Bakheit AH, Saquib Q, Ahmed S, Ansari SM, Al-Salem AM, Al-Khedhairy AA. Covalent Inhibitors from Saudi Medicinal Plants Target RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2. Viruses. 2023; 15(11):2175. https://doi.org/10.3390/v15112175
Chicago/Turabian StyleBakheit, Ahmed H., Quaiser Saquib, Sarfaraz Ahmed, Sabiha M. Ansari, Abdullah M. Al-Salem, and Abdulaziz A. Al-Khedhairy. 2023. "Covalent Inhibitors from Saudi Medicinal Plants Target RNA-Dependent RNA Polymerase (RdRp) of SARS-CoV-2" Viruses 15, no. 11: 2175. https://doi.org/10.3390/v15112175