
A Method to Generate and Rescue Recombinant Adenovirus Devoid of Replication-Competent Particles in Animal-Origin-Free Culture Medium

Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells Lines, Plasmids, and Viruses
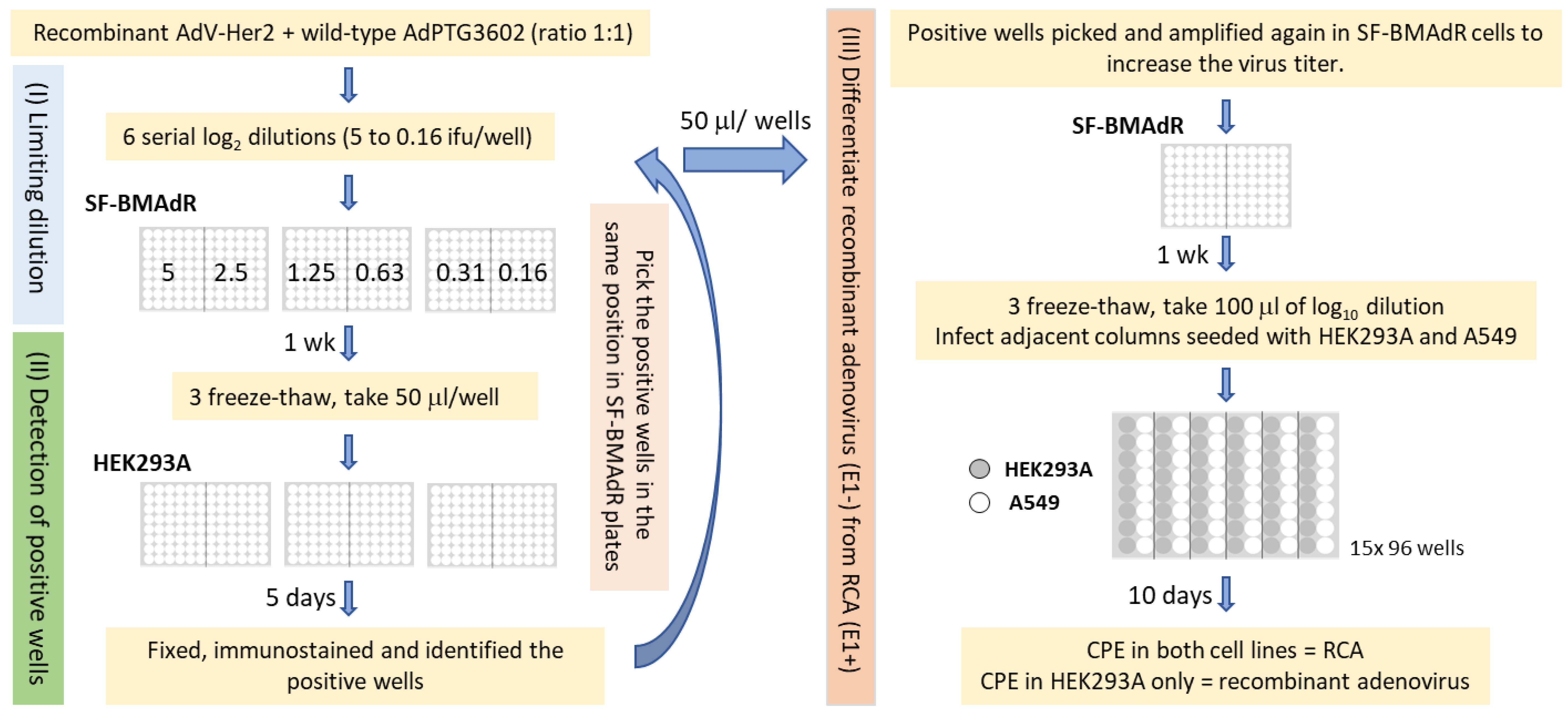
2.2. Developing a Limiting Dilution Method to Replace Plaque Purification
2.3. Generation of rAdV without FBS
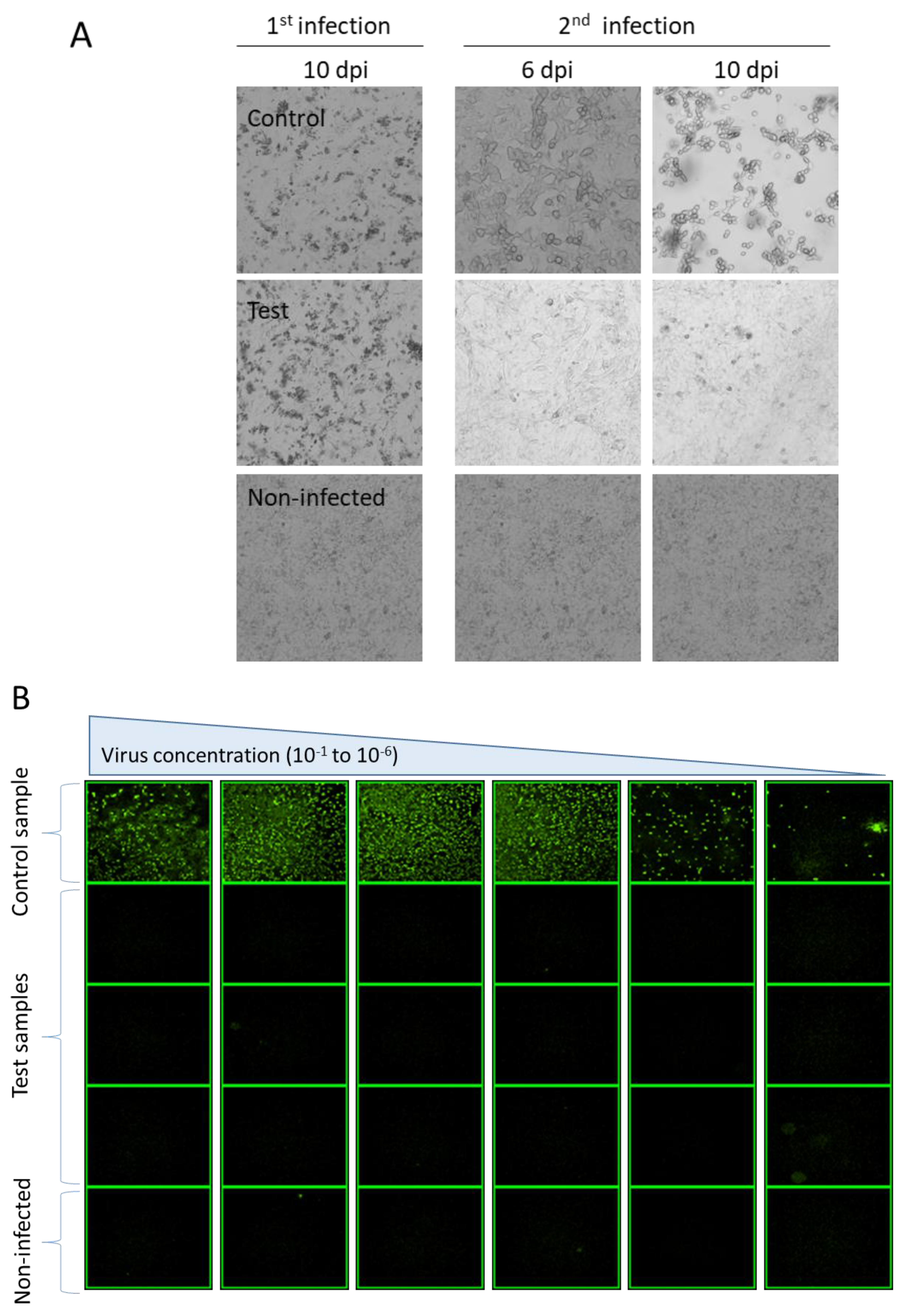
2.4. Scale-Up and RCA Detection
3. Results
3.1. Cloning AdV by Limiting Dilution
3.2. Generation of rAdV in the Absence of FBS and RCA
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Danthinne, X.; Imperiale, M.J. Production of First Generation Adenovirus Vectors: A Review. Gene Ther. 2000, 7, 1707–1714. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, R.F.; Kreppel, F. Production, Purification, and Titration of First-Generation Adenovirus Vectors. Methods Mol. Biol. 2017, 1654, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Tsilingiris, D.; Vallianou, N.G.; Karampela, I.; Muscogiuri, G.; Dalamaga, M. Use of Adenovirus Type-5 Vector Vaccines in COVID-19: Potential Implications for Metabolic Health? Minerva Endocrinol. 2022, 47, 264–269. [Google Scholar] [CrossRef]
- World Health Organization. COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 7 June 2023).
- Graham, F.L.; Smiley, J.; Russell, W.C.; Nairn, R. Characteristics of a Human Cell Line Transformed by DNA from Human Adenovirus Type 5. J. Gen. Virol. 1977, 36, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Louis, N.; Evelegh, C.; Graham, F.L. Cloning and Sequencing of the Cellular-Viral Junctions from the Human Adenovirus Type 5 Transformed 293 Cell Line. Virology 1997, 233, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Lochmüller, H.; Jani, A.; Huard, J.; Prescott, S.; Simoneau, M.; Massie, B.; Karpati, G.; Acsadi, G. Emergence of Early Region 1-Containing Replication-Competent Adenovirus in Stocks of Replication-Defective Adenovirus Recombinants (Delta E1 + Delta E3) during Multiple Passages in 293 Cells. Hum. Gene Ther. 1994, 5, 1485–1491. [Google Scholar] [CrossRef]
- Hehir, K.M.; Armentano, D.; Cardoza, L.M.; Choquette, T.L.; Berthelette, P.B.; White, G.A.; Couture, L.A.; Everton, M.B.; Keegan, J.; Martin, J.M.; et al. Molecular Characterization of Replication-Competent Variants of Adenovirus Vectors and Genome Modifications to Prevent Their Occurrence. J. Virol. 1996, 70, 8459–8467. [Google Scholar] [CrossRef]
- Zhu, J.; Grace, M.; Casale, J.; Chang, A.T.; Musco, M.L.; Bordens, R.; Greenberg, R.; Schaefer, E.; Indelicato, S.R. Characterization of Replication-Competent Adenovirus Isolates from Large-Scale Production of a Recombinant Adenoviral Vector. Hum. Gene Ther. 1999, 10, 113–121. [Google Scholar] [CrossRef]
- Food and Drug Administration. Chemistry, Manufacturing, and Control (CMC) Information for Human Gene Therapy Investigational New Drug Applications (INDs) Guidance for Industry; Food and Drug Administration: Silver Spring, MD, USA, 2020.
- Farson, D.; Tao, L.; Ko, D.; Li, Q.; Brignetti, D.; Segawa, K.; Mittelstaedt, D.; Harding, T.; Yu, D.-C.; Li, Y. Development of Novel E1-Complementary Cells for Adenoviral Production Free of Replication-Competent Adenovirus. Mol. Ther. 2006, 14, 305–311. [Google Scholar] [CrossRef]
- Gilbert, R.; Guilbault, C.; Gagnon, D.; Bernier, A.; Bourget, L.; Elahi, S.M.; Kamen, A.; Massie, B. Establishment and Validation of New Complementing Cells for Production of E1-Deleted Adenovirus Vectors in Serum-Free Suspension Culture. J. Virol. Methods 2014, 208, 177–188. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, S.H.; Cho, Y.S.; Park, K.; Kim, Y.H.; Lee, J.H. Development of a Packaging Cell Line for Propagation of Replication-Deficient Adenovirus Vector. Exp. Mol. Med. 2001, 33, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Kovesdi, I.; Hedley, S.J. Adenoviral Producer Cells. Viruses 2010, 2, 1681–1703. [Google Scholar] [CrossRef] [PubMed]
- Howe, J.A.; Pelka, P.; Antelman, D.; Wilson, C.; Cornell, D.; Hancock, W.; Ramachandra, M.; Avanzini, J.; Horn, M.; Wills, K.; et al. Matching Complementing Functions of Transformed Cells with Stable Expression of Selected Viral Genes for Production of E1-Deleted Adenovirus Vectors. Virology 2006, 345, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Arevalo, M.T.; Pichichero, M.E.; Zeng, M. A New Complementing Cell Line for Replication-Incompetent E1-Deleted Adenovirus Propagation. Cytotechnology 2006, 51, 133–140. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New Helper Cells and Matched Early Region 1-Deleted Adenovirus Vectors Prevent Generation of Replication-Competent Adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef]
- Gao, G.P.; Engdahl, R.K.; Wilson, J.M. A Cell Line for High-Yield Production of E1-Deleted Adenovirus Vectors without the Emergence of Replication-Competent Virus. Hum. Gene Ther. 2000, 11, 213–219. [Google Scholar] [CrossRef]
- Xie, L.; Pilbrough, W.; Metallo, C.; Zhong, T.; Pikus, L.; Leung, J.; Auniņs, J.G.; Zhou, W. Serum-Free Suspension Cultivation of PER.C6(R) Cells and Recombinant Adenovirus Production under Different PH Conditions. Biotechnol. Bioeng. 2002, 80, 569–579. [Google Scholar] [CrossRef]
- Gstraunthaler, G.; Lindl, T.; van der Valk, J. A Plea to Reduce or Replace Fetal Bovine Serum in Cell Culture Media. Cytotechnology 2013, 65, 791–793. [Google Scholar] [CrossRef]
- Young, K.G.; Haq, K.; MacLean, S.; Dudani, R.; Elahi, S.M.; Gilbert, R.; Weeratna, R.D.; Krishnan, L. Development of a Recombinant Murine Tumour Model Using Hepatoma Cells Expressing Hepatitis C Virus Nonstructural Antigens. J. Viral. Hepat. 2018, 25, 649–660. [Google Scholar] [CrossRef]
- Haq, K.; Jia, Y.; Elahi, S.M.; MacLean, S.; Akache, B.; Gurnani, K.; Chattopadhyay, A.; Nazemi-Moghaddam, N.; Gilbert, R.; McCluskie, M.J.; et al. Evaluation of Recombinant Adenovirus Vectors and Adjuvanted Protein as a Heterologous Prime-Boost Strategy Using HER2 as a Model Antigen. Vaccine 2019, 37, 7029–7040. [Google Scholar] [CrossRef]
- Oualikene, W.; Lamoureux, L.; Weber, J.M.; Massie, B. Protease-Deleted Adenovirus Vectors and Complementing Cell Lines: Potential Applications of Single-Round Replication Mutants for Vaccination and Gene Therapy. Hum. Gene Ther. 2000, 11, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.M.; Nazemi-Moghaddam, N.; Gadoury, C.; Lippens, J.; Radinovic, S.; Venne, M.-H.; Marcil, A.; Gilbert, R. A Rapid Focus-Forming Assay for Quantification of Infectious Adenoviral Vectors. J. Virol. Methods 2021, 297, 114267. [Google Scholar] [CrossRef] [PubMed]
- Mittereder, N.; March, K.L.; Trapnell, B.C. Evaluation of the Concentration and Bioactivity of Adenovirus Vectors for Gene Therapy. J. Virol. 1996, 70, 7498–7509. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.S.; Ganesh, S.; Hawkins, L.; Idamakanti, N. Generation of Recombinant Adenovirus Using the Escherichia Coli BJ5183 Recombination System. Methods Mol. Med. 2007, 130, 61–68. [Google Scholar] [CrossRef]
- Søndergaard, J.N.; Geng, K.; Sommerauer, C.; Atanasoai, I.; Yin, X.; Kutter, C. Successful Delivery of Large-Size CRISPR/Cas9 Vectors in Hard-to-Transfect Human Cells Using Small Plasmids. Commun. Biol. 2020, 3, 319. [Google Scholar] [CrossRef] [PubMed]
- Murakami, P.; Havenga, M.; Fawaz, F.; Vogels, R.; Marzio, G.; Pungor, E.; Files, J.; Do, L.; Goudsmit, J.; McCaman, M. Common Structure of Rare Replication-Deficient E1-Positive Particles in Adenoviral Vector Batches. J. Virol. 2004, 78, 6200–6208. [Google Scholar] [CrossRef]
- Schalk, J.A.C.; de Vries, C.G.J.C.A.; Orzechowski, T.J.H.; Rots, M.G. A Rapid and Sensitive Assay for Detection of Replication-Competent Adenoviruses by a Combination of Microcarrier Cell Culture and Quantitative PCR. J. Virol. Methods 2007, 145, 89–95. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Infectious Dose (ifu)/Well (SF-BMAdR) | 5 | 2.5 | 1.25 | 0.63 | 0.31 | 0.16 | Total of All Wells |
|---|---|---|---|---|---|---|---|
| Total positive wells in FFA (HEK293A) | 38 | 27 | 14 | 7 | 5 | 1 | 92 |
| Amplifed wells in SF-BMAdR cells | 35 | 27 | 14 | 7 | 5 | 1 | 89 |
| RCA (amplified in HEK293A & A549 cells) | 16 | 10 | 7 | 2 | 0 | 0 | 35 |
| AdV (amplified only in HEK293A) | 13 | 11 | 6 | 3 | 5 | 1 | 39 |
| Exclueded (Not amplified or had low titer) | 6 | 6 | 1 | 2 | 0 | 0 | 15 |
| Cell Lines | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HEK-293A | A549 | HEK-293A | A549 | HEK-293A | A549 | HEK-293A | A549 | HEK-293A | A549 | HEK-293A | A549 | |
| #1 | #2 | #3 | #4 | #5 | #6 | |||||||
| Log10 dilution 10−1 to 10−8 | + | + | + | + | + | − | + | + | + | − | − | − |
| + | + | + | + | + | − | + | − | − | − | − | − | |
| + | + | + | + | + | − | + | − | − | − | − | − | |
| + | + | − | − | + | − | + | − | − | − | − | − | |
| + | + | − | − | + | − | + | − | − | − | − | − | |
| + | + | − | − | − | − | + | − | − | − | − | − | |
| + | − | − | − | − | − | − | − | − | − | − | − | |
| − | − | − | − | − | − | − | − | − | − | − | − | |
| Interpretation | ||||||||||||
| RCA | RCA | AdV | AdV | Excluded | Excluded | |||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elahi, S.M.; Jiang, J.; Nazemi-Moghaddam, N.; Gilbert, R. A Method to Generate and Rescue Recombinant Adenovirus Devoid of Replication-Competent Particles in Animal-Origin-Free Culture Medium. Viruses 2023, 15, 2152. https://doi.org/10.3390/v15112152
Elahi SM, Jiang J, Nazemi-Moghaddam N, Gilbert R. A Method to Generate and Rescue Recombinant Adenovirus Devoid of Replication-Competent Particles in Animal-Origin-Free Culture Medium. Viruses. 2023; 15(11):2152. https://doi.org/10.3390/v15112152
Chicago/Turabian StyleElahi, Seyyed Mehdy, Jennifer Jiang, Nazila Nazemi-Moghaddam, and Rénald Gilbert. 2023. "A Method to Generate and Rescue Recombinant Adenovirus Devoid of Replication-Competent Particles in Animal-Origin-Free Culture Medium" Viruses 15, no. 11: 2152. https://doi.org/10.3390/v15112152