
Unraveling DPP4 Receptor Interactions with SARS-CoV-2 Variants and MERS-CoV: Insights into Pulmonary Disorders via Immunoinformatics and Molecular Dynamics
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Docking Studies
2.2. System Preparation
2.3. MD Simulation Studies
2.4. Protein Stability and Flexibility Analysis
2.5. Perturbation Residue Scanning (PRS)
3. Results
3.1. Comparative Analysis of Spike Variants and DPP4 Interaction
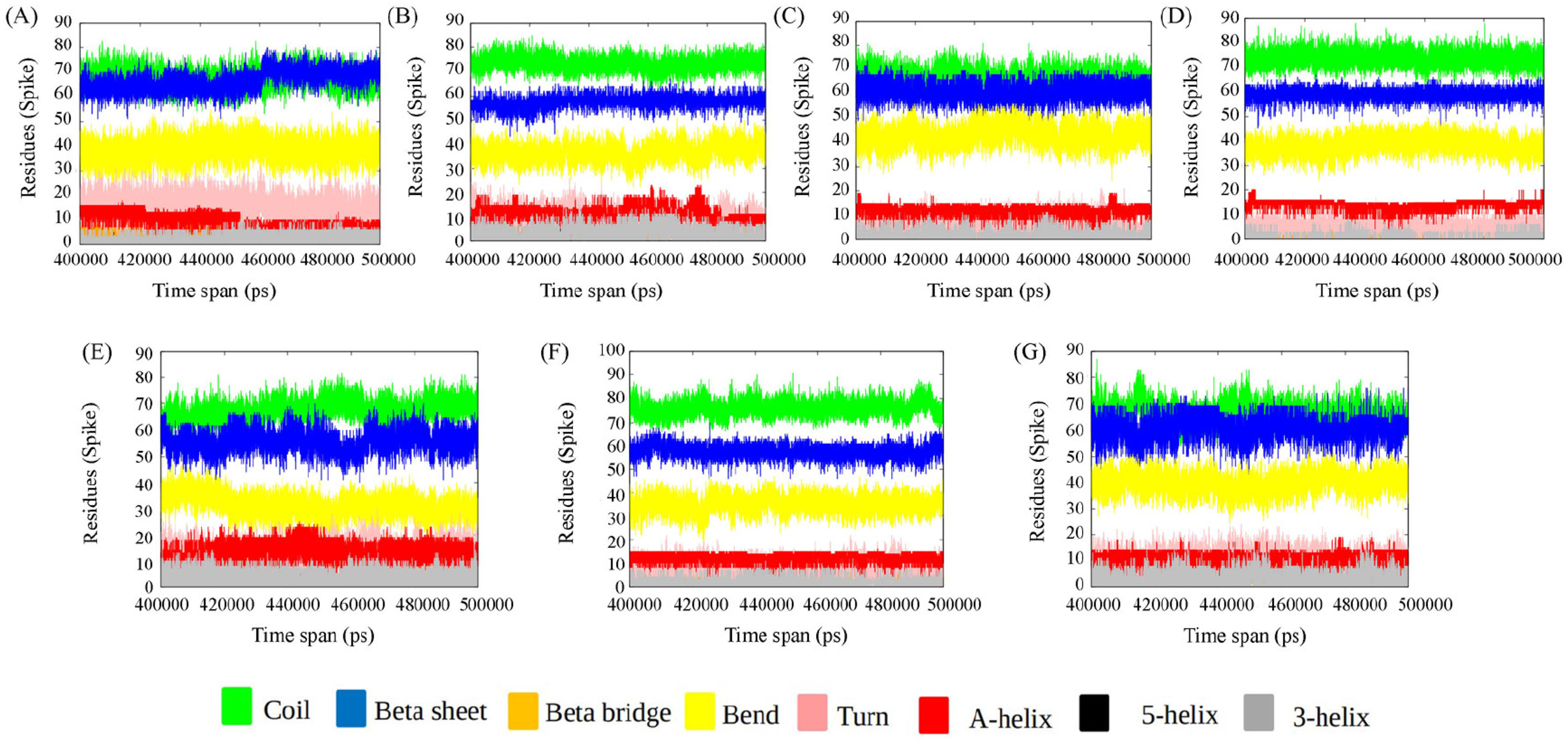
3.1.1. Secondary Structure
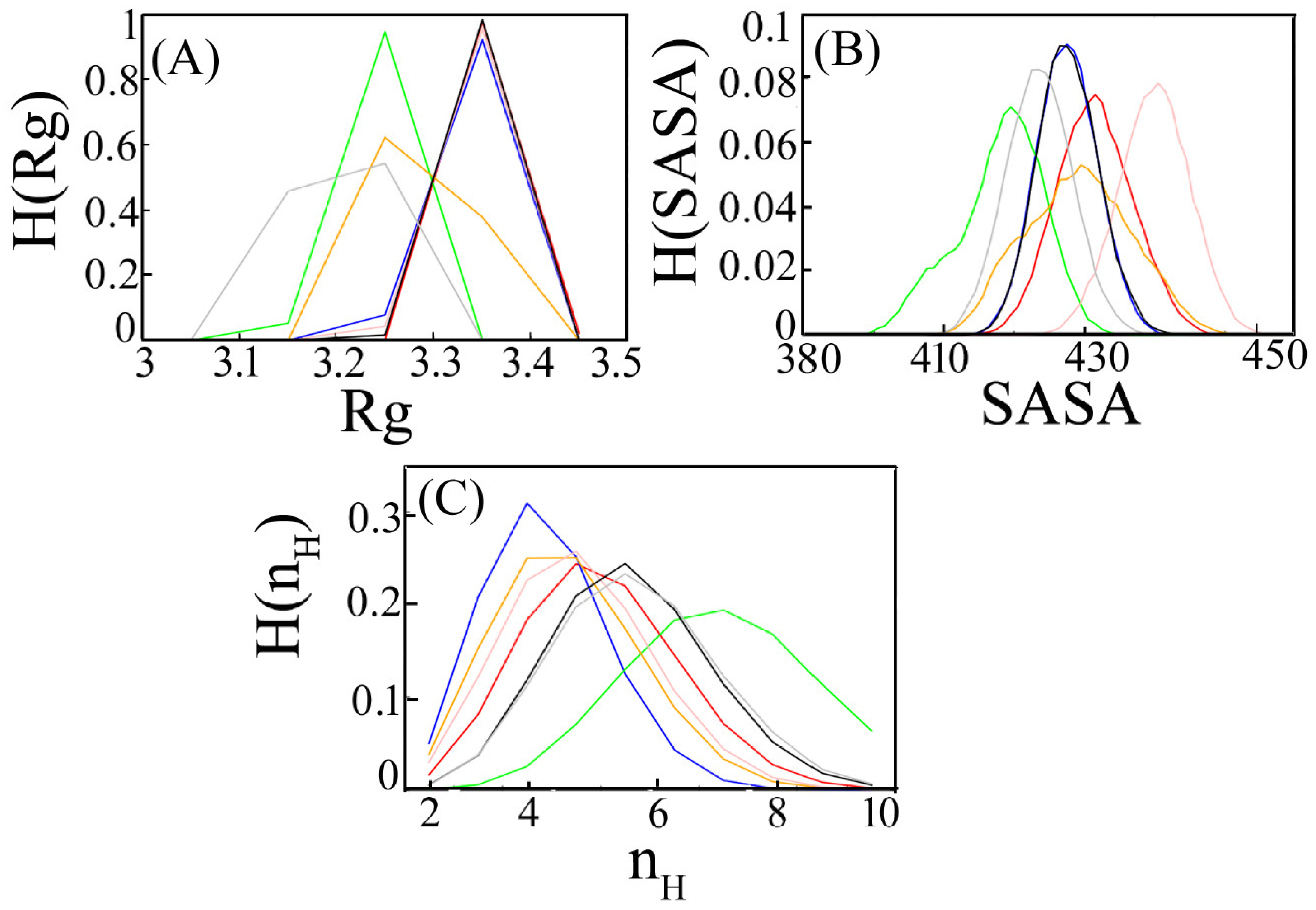
3.1.2. Radius of Gyration
3.1.3. Accessible Surface Area
3.1.4. Hydrogen Bonds
3.1.5. Interface Analysis
3.2. Fluctuations in the Complexes
3.2.1. RMSF
3.2.2. PCA
3.2.3. Protein Stability
3.2.4. Perturbation Residue Scanning (PRS)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matteucci, E.; Giampietro, O. Dipeptidyl peptidase-4 (CD26): Knowing the function before inhibiting the enzyme. Curr. Med. Chem. 2009, 16, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Ulmer, A.J.; Mattern, T.; Feller, A.C.; Heymann, E.; Flad, H.-D. CD26 antigen is a surface dipeptidyl peptidase IV (DPPIV) as characterized by monoclonal antibodies clone til-19-4-7 and 4EL1C7. Scand. J. Immunol. 1990, 31, 429–435. [Google Scholar] [CrossRef]
- Morrison, M.E.; Vijayasaradhi, S.; Engelstein, D.; Albino, A.P.; Houghton, A.N. A marker for neoplastic progression of human melanocytes is a cell surface ectopeptidase. J. Exp. Med. 1993, 177, 1135–1143. [Google Scholar] [CrossRef]
- Fleischer, B. CD26: A surface protease involved in T-cell activation. Immunol. Today 1994, 15, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Van Der, V.; Hulsmann, A.R. Peptidases: Structure, function and modulation of peptide-mediated effects in the human lung. Clin. Amp; Exp. Allergy 1999, 29, 445–456. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.; Yang, L.; Lian, X.; Xie, Y.; Li, S.; Xin, S.; Cao, P.; Lu, J. The MERS-COV receptor DPP4 as a candidate binding target of the SARS-COV-2 spike. iScience 2020, 23, 101160. [Google Scholar] [CrossRef] [PubMed]
- Vankadari, N.; Wilce, J.A. Emerging COVID-19 coronavirus: Glycan Shield and structure prediction of Spike glycoprotein and its interaction with human CD26. Emerg. Microbes Amp Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Cameron, K.; Rozano, L.; Falasca, M.; Mancera, R.L. Does the SARS-CoV-2 spike protein receptor binding domain interact effectively with the DPP4 (CD26) receptor? A molecular docking study. Int. J. Mol. Sci. 2021, 22, 7001. [Google Scholar] [CrossRef]
- Chan, C.-M.; Chu, H.; Wang, Y.; Wong, B.H.; Zhao, X.; Zhou, J.; Yang, D.; Leung, S.P.; Chan, J.F.; Yeung, M.L.; et al. Carcinoembryonic antigen-related cell adhesion molecule 5 is an important surface attachment factor that facilitates entry of Middle East respiratory syndrome coronavirus. J. Virol. 2016, 90, 9114–9127. [Google Scholar] [CrossRef]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef]
- Cheng, F.; Yuan, G.; He, J.; Shao, Y.; Zhang, J.; Guo, X. Dysregulation of DPP4 is associated with the AMPK/jak2/STAT3 pathway in adipocytes under insulin resistance status and liraglutide intervention. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2635–2644. [Google Scholar] [CrossRef]
- Millet, J.K.; Jaimes, A.J.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2020, 45, fuaa057. [Google Scholar] [CrossRef]
- Earnest, J.T.; Hantak, M.P.; Li, K.; McCray, P.B., Jr.; Perlman, S.; Gallagher, T. The tetraspanin CD9 facilitates mers-coronavirus entry by scaffolding host cell receptors and proteases. PLoS Pathog. 2017, 13, e1006546. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-COV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Guo, D.; Fu, L.; Cui, Y.; Liu, X.; et al. Structure of MERS-COV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, Y.; Wang, N.; Wang, D.; Guo, J.; Fu, L.; Shi, X. Identification of residues on human receptor DPP4 critical for MERS-COV binding and entry. Virology 2014, 471–473, 49–53. [Google Scholar] [CrossRef]
- Wei, L.; Ming, S.; Zou, B.; Wu, Y.; Hong, Z.; Li, Z.; Zheng, X.; Huang, M.; Luo, L.; Liang, J.; et al. Viral invasion and type I interferon response characterize the immunophenotypes during COVID-19 infection. SSRN Electron. J. 2020, 1–27. [Google Scholar] [CrossRef]
- Solerte, S.B.; Di Sabatino, A.; Galli, M.; Fiorina, P. Dipeptidyl peptidase-4 (DPP4) inhibition in COVID-19. Acta Diabetol. 2020, 57, 779–783. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-ncov infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Yang, J.; Zheng, Y.; Gou, X.; Pu, K.; Chen, Z.; Guo, Q.; Ji, R.; Wang, H.; Wang, Y.; Zhou, Y. Prevalence of comorbidities and its effects in patients infected with SARS-CoV-2: A systematic review and meta-analysis. Int. J. Infect. Dis. 2020, 94, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Nistala, R.; Savin, V. Diabetes, hypertension, and chronic kidney disease progression: Role of DPP4. Am. J. Physiol.-Ren. Physiol. 2017, 312, F661–F670. [Google Scholar] [CrossRef]
- Rohmann, N.; Schlicht, K.; Geisler, C.; Hollstein, T.; Knappe, C.; Krause, L.; Hagen, S.; Beckmann, A.; Seoudy, A.K.; Wietzke-Braun, P.; et al. Circulating SDPP-4 is increased in obesity and insulin resistance but is not related to systemic metabolic inflammation. J. Clin. Endocrinol. Amp Metab. 2020, 106, e592–e601. [Google Scholar] [CrossRef] [PubMed]
- Barchetta, I.; Ceccarelli, V.; Cimini, F.A.; Barone, E.; Sentinelli, F.; Coluzzi, M.; Chiappetta, C.; Bertoccini, L.; Tramutola, A.; Labbadia, G.; et al. Circulating dipeptidyl peptidase-4 is independently associated with the presence and severity of NAFLD/Nash in individuals with and without obesity and metabolic disease. J. Endocrinol. Investig. 2020, 44, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.J.; Lamarche, B.; Deacon, C.F.; Weisnagel, S.J.; Couture, P. Effect of sitagliptin therapy on postprandial lipoprotein levels in patients with type 2 diabetes. Diabetes Obes. Metab. 2011, 13, 366–373. [Google Scholar] [CrossRef]
- Metzemaekers, M.; Van Damme, J.; Mortier, A.; Proost, P. Regulation of chemokine activity—A focus on the role of dipeptidyl peptidase IV/CD26. Front. Immunol. 2016, 7, 483. [Google Scholar] [CrossRef]
- Portelli, M.; Sayers, I. Genetic basis for personalized medicine in asthma. Expert Rev. Respir. Med. 2012, 6, 223–236. [Google Scholar] [CrossRef]
- Mathias, R.A.; Grant, A.V.; Rafaels, N.; Hand, T.; Gao, L.; Vergara, C.; Tsai, Y.J.; Yang, M.; Campbell, M.; Foster, C.; et al. A genome-wide association study on African-ancestry populations for asthma. J. Allergy Clin. Immunol. 2010, 125, 336–346.e4. [Google Scholar] [CrossRef]
- Mitchell, J.; Dimov, V.; Townley, R.G. IL-13 and the IL-13 receptor as therapeutic targets for asthma and allergic disease. Curr Opin Investig Drugs 2010, 11, 527–534. [Google Scholar] [PubMed]
- Anderluh, M.; Kocic, G.; Tomovic, K.; Kocic, H.; Smelcerovic, A. DPP-4 inhibition: A novel therapeutic approach to the treatment of pulmonary hypertension? Pharmacol. Amp Ther. 2019, 201, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, J.; He, M.; Han, H.; Xie, W.; Wang, H.; Kong, H. Dipeptidyl peptidase IV (DPP-4) inhibition alleviates pulmonary arterial remodeling in experimental pulmonary hypertension. Lab. Investig. 2018, 98, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Laird, M.; Duffy, D.; Casrouge, A.; Mamdouh, R.; Abass, A.; El Shenawy, D.; Shebl, A.M.; Elkashef, W.F.; Zalata, K.R.; et al. Corrigendum to “CXCL10 antagonism and plasma SDPPIV correlate with increasing liver disease in chronic HCV genotype 4 infected patients” [CYTOKINE 63 (2013) 105–112]. Cytokine 2014, 65, 119–120. [Google Scholar] [CrossRef]
- Casrouge, A.; Decalf, J.; Ahloulay, M.; Lababidi, C.; Mansour, H.; Vallet-Pichard, A.; Mallet, V.; Mottez, E.; Mapes, J.; Fontanet, A.; et al. Evidence for an antagonist form of the chemokine CXCL10 in patients chronically infected with HCV. J. Clin. Investig. 2011, 121, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Rainczuk, A.; Rao, J.R.; Gathercole, J.L.; Fairweather, N.J.; Chu, S.; Masadah, R.; Jobling, T.W.; Deb-Choudhury, S.; Dyer, J.; Stephens, A.N. Evidence for the antagonistic form of CXC-motif chemokine CXCL10 in serous epithelial ovarian tumours. Int. J. Cancer 2013, 134, 530–541. [Google Scholar] [CrossRef] [PubMed]
- Blauenfeldt, T.; Petrone, L.; Del Nonno, F.; Baiocchini, A.; Falasca, L.; Chiacchio, T.; Bondet, V.; Vanini, V.; Palmieri, F.; Galluccio, G.; et al. Interplay of DDP4 and IP-10 as a potential mechanism for cell recruitment to tuberculosis lesions. Front. Immunol. 2018, 9, 1456. [Google Scholar] [CrossRef]
- de Vries, S.J.; van Dijk, M.; Bonvin, A.M. The haddock web server for data-driven biomolecular docking. Nat. Protoc. 2010, 5, 883–897. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-COV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.P.; van Gunsteren, W.F.; Hermans, J. Interaction models for water in relation to protein hydration. In The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Springer: Dordrecht, The Netherlands, 1981; pp. 331–342. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The gromos force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An n⋅log(n) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B. Dynamut: Predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res. 2018, 46, W350–W355. [Google Scholar] [CrossRef]
- Gopi, S.; Devanshu, D.; Rajasekaran, N.; Anantakrishnan, S.; Naganathan, A.N. PPerturb: A server for predicting long-distance energetic couplings and mutation-induced stability changes in proteins via perturbations. ACS Omega 2020, 5, 1142–1146. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Suresh, S.; Gopi, S.; Raman, K.; Naganathan, A.N. A general mechanism for the propagation of mutational effects in proteins. Biochemistry 2016, 56, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, L.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, Y.; Zhou, Y.; Zhang, J.; Zhang, Z. HADDOCK-based prediction of PD-1/PD-L1 interaction and the identification of potential drug targets. Front. Immunol. 2019, 10, 1558. [Google Scholar]
- Dominguez, C.; Rueda, M.; Fiore, A.; Bonvin, A.M.J.J. HADDOCK docking of HIV-1 gp120 to CD4: Implications for the design of gp120 mimics. Protein Sci. 2005, 14, 435–442. [Google Scholar]
- Guharoy, M.; Chakrabarti, P. Secondary structure based analysis and classification of biological interfaces: Identification of binding motifs in protein–protein interactions. Bioinformatics 2007, 23, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.A.; Teichmann, S.A. Protein flexibility facilitates quaternary structure assembly and Evolution. PLoS Biol. 2014, 12, e1001870. [Google Scholar] [CrossRef]
- Jubb, H.; Blundell, T.L.; Ascher, D.B. Flexibility and small pockets at protein–protein interfaces: New insights into druggability. Prog. Biophys. Mol. Biol. 2015, 119, 2–9. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Dynamics of proteins and nucleic acids. Annu. Rev. Biophys. Bioeng. 1983, 12, 263–300. [Google Scholar]
- Amadei, A.; Linssen, A.B.M.; Berendsen, H.J.C. Essential dynamics of proteins. Proteins Struct. Funct. Bioinform. 1993, 17, 412–425. [Google Scholar] [CrossRef]
- Rajasekaran, N.; Naganathan, A.N. A self-consistent structural perturbation approach for determining the magnitude and extent of allosteric coupling in proteins. Biochem. J. 2017, 474, 2379–2388. [Google Scholar] [CrossRef] [PubMed]
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.-J.; Walls, A.C.; Wang, Z.; Sauer, M.M.; Li, W.; Tortorici, M.A.; Bosch, B.J.; DiMaio, F.; Veesler, D. Structures of MERS-cov spike glycoprotein in complex with sialoside attachment receptors. Nat. Struct. Amp Mol. Biol. 2019, 26, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Schleker, S.; Garcia-Garcia, J.; Klein-Seetharaman, J.; Oliva, B. Prediction and comparison of salmonella-human and salmonella-arabidopsis interactomes. Chem. Biodivers. 2012, 9, 991–1018. [Google Scholar] [CrossRef]
- von Mering, C. String: Known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2004, 33, D433–D437. [Google Scholar] [CrossRef]
- Yang, X.; Yang, S.; Li, Q.; Wuchty, S.; Zhang, Z. Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method. Comput. Struct. Biotechnol. J. 2020, 18, 153–161. [Google Scholar] [CrossRef]
- Zhou, H.; Gao, S.; Nguyen, N.N.; Fan, M.; Jin, J.; Liu, B.; Zhao, L.; Xiong, G.; Tan, M.; Li, S.; et al. Stringent homology-based prediction of H. Sapiens-M. tuberculosis H37RV protein-protein interactions. Biol. Direct 2014, 9, 5. [Google Scholar] [CrossRef]
- Ahmed, H.; Patel, K.; Greenwood, D.C.; Halpin, S.; Lewthwaite, P.; Salawu, A.; Eyre, L.; Breen, A.; O’Connor, R.; Jones, A.; et al. Long-term clinical outcomes in survivors of severe acute respiratory syndrome and Middle East respiratory syndrome coronavirus outbreaks after hospitalisation or ICU admission: A systematic review and meta-analysis. J. Rehabil. Med. 2020, 52, jrm00063. [Google Scholar] [CrossRef]
- Hui, D.S.; Joynt, G.M.; Wong, K.T.; Gomersall, C.D.; Li, T.S.; Antonio, G.; Ko, F.W.; Chan, M.C.; Chan, D.P.; Tong, M.W.; et al. Impact of severe acute respiratory syndrome (SARS) on pulmonary function, functional capacity, and quality of life in a cohort of survivors. Thorax 2005, 60, 401–409. [Google Scholar] [CrossRef]
- Lam, M.H.; Wing, Y.K.; Yu, M.W.; Leung, C.M.; Ma, R.C.; Kong, A.P.; So, W.Y.; Fong, S.Y.; Lam, S.P. Mental morbidities and chronic fatigue in severe acute respiratory syndrome survivors: Long-term follow-up. Arch. Intern. Med. 2009, 169, 2142–2147. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Rg (nm) | SASA (nm/S2/N) | nH (no.) |
|---|---|---|---|
| MERS CoV:DPP4 | 3.200 | 4.135 | 4.208 |
| WT-spike SARS CoV-2:DPP4 | 3.340 | 4.176 | 4.102 |
| Alpha SARS CoV-2:DPP4 | 3.358 | 4.214 | 3.494 |
| Beta SARS CoV-2:DPP4 | 3.328 | 4.175 | 2.375 |
| Delta SARS CoV-2:DPP4 | 3.239 | 4.079 | 5.998 |
| Gamma SARS CoV-2:DPP4 | 3.289 | 4.191 | 2.832 |
| Omicron SARS CoV-2:DPP4 | 3.344 | 4.299 | 3.057 |
| Delta | Wildtype | Gamma | Alpha | Beta | Omicron | MERS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | SARS-CoV-2 Residue | DPP4 Residue | MERS Residue |
| A282 A289 A291 K287 D331 K392 E191 R336 K190 E347 E332 D390 D192 R343 S284 S284 S284 Q344 Y225 E191 | V483 Q493 Q493 T478 R452 D442 K478 E471 E484 K444 R452 K444 K478 D442 C488 N487 G485 G504 N487 S477 | A282 A289 A291 F269 I285 A291 A342 Q286 D331 R336 K392 D393 R343 | V483 Q493 Q493 V483 V483 Y495 N448 E484 S494 E484 D442 K444 D442 | A282 A289 A291 F269 I285 I287 Q286 Q444 E332 D393 A342 | V483 Q493 Q493 V483 V483 V483 N481 K444 K484 K444 N448 | A282 A289 F269 I485 I287 Q344 K392 R336 S284 Q286 A342 | V483 Q493 V483 V483 V483 K444 D442 E471 K484 K484 N450 | A289 A291 A291 I285 E347 E378 D393 A291 L294 S292 | Q493 F456 Q493 V483 K444 K444 K444 Y495 G502 G504 | A282 A289 Q344 T288 D390 N281 A282 | V483 Q493 K444 Y505 K444 C488 N487 | K267 R317 R336 L294 A289 A291 L294 L294 | D539 D510 Y499 R542 K502 L506 Y540 V555 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, A.N.; Gupta, A.M.; Banerjee, D.; Chakrabarti, J.; Raghavendra, P.B. Unraveling DPP4 Receptor Interactions with SARS-CoV-2 Variants and MERS-CoV: Insights into Pulmonary Disorders via Immunoinformatics and Molecular Dynamics. Viruses 2023, 15, 2056. https://doi.org/10.3390/v15102056
Roy AN, Gupta AM, Banerjee D, Chakrabarti J, Raghavendra PB. Unraveling DPP4 Receptor Interactions with SARS-CoV-2 Variants and MERS-CoV: Insights into Pulmonary Disorders via Immunoinformatics and Molecular Dynamics. Viruses. 2023; 15(10):2056. https://doi.org/10.3390/v15102056
Chicago/Turabian StyleRoy, Arpan Narayan, Aayatti Mallick Gupta, Deboshmita Banerjee, Jaydeb Chakrabarti, and Pongali B. Raghavendra. 2023. "Unraveling DPP4 Receptor Interactions with SARS-CoV-2 Variants and MERS-CoV: Insights into Pulmonary Disorders via Immunoinformatics and Molecular Dynamics" Viruses 15, no. 10: 2056. https://doi.org/10.3390/v15102056