

RNA Editing-Dependent and -Independent Roles of Adenosine Deaminases Acting on RNA Proteins in Herpesvirus Infection—Hints on Another Layer of Complexity

Abstract
:1. Introduction
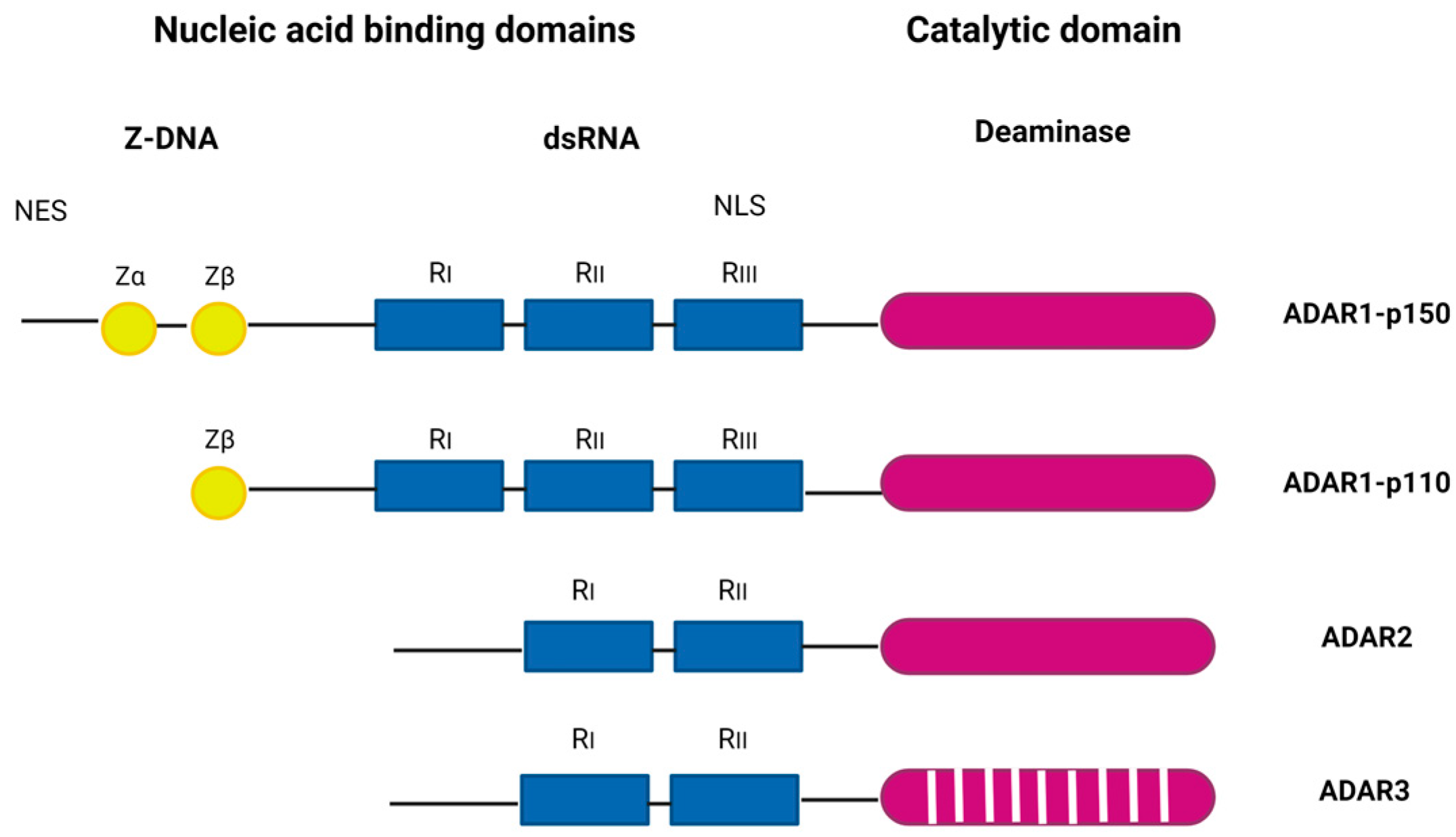
2. The ADAR Protein Family in Brief
3. Herpesviruses in Brief
The Orthoherpesviridae
4. ADAR and Herpesviruses
4.1. Evidence for ADAR-Mediated RNA Editing in Alphaherpesviruses
4.1.1. Herpes Simplex Virus 1 (HSV-1)—Latent miRNA Editing
4.1.2. Varicella Zoster Virus (VZV)—Edited Novel Viral Transcripts
4.1.3. Gallid Herpesvirus 2 (GaHV-2, or Marek’s Disease Virus 1 (MDV-1)
4.2. Evidence of ADAR-Mediated RNA Editing in Betaherpesviruses—Edited Host miRNA
4.3. Gammaherpesviruses: Editing-Dependent and Editing-Independent Roles of ADAR Proteins
4.3.1. Epstein–Barr Virus (EBV, HHV-4)—Editing Affects miRNA Biogenesis
4.3.2. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV, HHV-8)
- KSHV—RNA Editing Phenomena
- KSHV—Editing-Independent Roles of ADAR
4.4. The Malacoherpesviridae–Herpesviruses of Mollusks
5. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Vukic, D.; Michalik, D.; O’Connell, M.A.; Keegan, L.P. ADAR RNA editing in human disease; more to it than meets the I. Hum. Genet. 2017, 136, 1265–1278. [Google Scholar] [CrossRef]
- Tomaselli, S.; Bonamassa, B.; Alisi, A.; Nobili, V.; Locatelli, F.; Gallo, A. ADAR enzyme and miRNA story: A nucleotide that can make the difference. Int. J. Mol. Sci. 2013, 14, 22796–22816. [Google Scholar] [CrossRef] [PubMed]
- Tomaselli, S.; Galeano, F.; Locatelli, F.; Gallo, A. ADARs and the Balance Game between Virus Infection and Innate Immune Cell Response. Curr. Issues Mol. Biol. 2015, 17, 37–51. [Google Scholar]
- Samuel, C.E. Adenosine deaminase acting on RNA (ADAR1), a suppressor of double-stranded RNA-triggered innate immune responses. J. Biol. Chem. 2019, 294, 1710–1720. [Google Scholar] [CrossRef]
- Nishikura, K. A-to-I editing of coding and non-coding RNAs by ADARs. Nat. Rev. Mol. Cell Biol. 2016, 17, 83–96. [Google Scholar] [CrossRef]
- Bazak, L.; Haviv, A.; Barak, M.; Jacob-Hirsch, J.; Deng, P.; Zhang, R.; Isaacs, F.J.; Rechavi, G.; Li, J.B.; Eisenberg, E.; et al. A-to-I RNA editing occurs at over a hundred million genomic sites, located in a majority of human genes. Genome Res. 2014, 24, 365–376. [Google Scholar] [CrossRef]
- Peng, Z.; Cheng, Y.; Tan, B.C.; Kang, L.; Tian, Z.; Zhu, Y.; Zhang, W.; Liang, Y.; Hu, X.; Tan, X.; et al. Comprehensive analysis of RNA-Seq data reveals extensive RNA editing in a human transcriptome. Nat. Biotechnol. 2012, 30, 253–260. [Google Scholar] [CrossRef]
- Pfaller, C.K.; George, C.X.; Samuel, C.E. Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections. Annu. Rev. Virol. 2021, 8, 239–264. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Rosenberg, B.R.; Chung, H.; Rice, C.M. Identification of ADAR1 p150 and p110 Associated Edit Sites. Methods Mol. Biol. 2023, 2651, 285–294. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Zimmermann, A.; Oiknine Djian, E.; Weisblum, Y.; Livneh, Y.; Khanh Le, V.T.; Galun, E.; Horejsi, V.; Isakov, O.; Shomron, N.; et al. MicroRNA editing facilitates immune elimination of HCMV infected cells. PLoS Pathog. 2014, 10, e1003963. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposi’s sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLoS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef]
- Hood, J.L.; Morabito, M.V.; Martinez, C.R., 3rd; Gilbert, J.A.; Ferrick, E.A.; Ayers, G.D.; Chappell, J.D.; Dermody, T.S.; Emeson, R.B. Reovirus-mediated induction of ADAR1 (p150) minimally alters RNA editing patterns in discrete brain regions. Mol. Cell. Neurosci. 2014, 61, 97–109. [Google Scholar] [CrossRef]
- Wang, Q.; Miyakoda, M.; Yang, W.; Khillan, J.; Stachura, D.L.; Weiss, M.J.; Nishikura, K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004, 279, 4952–4961. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef]
- Herbert, A. Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zalpha domain of the double-stranded RNA editing enzyme ADAR. Eur. J. Hum. Genet. 2020, 28, 114–117. [Google Scholar] [CrossRef]
- Miyamura, Y.; Suzuki, T.; Kono, M.; Inagaki, K.; Ito, S.; Suzuki, N.; Tomita, Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 2003, 73, 693–699. [Google Scholar] [CrossRef]
- Baker, A.R.; Slack, F.J. ADAR1 and its implications in cancer development and treatment. Trends Genet. 2022, 38, 821–830. [Google Scholar] [CrossRef]
- Stok, J.E.; Oosenbrug, T.; Ter Haar, L.R.; Gravekamp, D.; Bromley, C.P.; Zelenay, S.; Reis e Sousa, C.; van der Veen, A.G. RNA sensing via the RIG-I-like receptor LGP2 is essential for the induction of a type I IFN response in ADAR1 deficiency. EMBO J. 2022, 41, e109760. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Deng, P.; Zhu, Z.; Zhu, J.; Wang, G.; Zhang, L.; Chen, A.F.; Wang, T.; Sarkar, S.N.; Billiar, T.R.; et al. Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs. J. Immunol. 2014, 193, 3436–3445. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.; Calis, J.J.A.; Wu, X.; Sun, T.; Yu, Y.; Sarbanes, S.L.; Dao Thi, V.L.; Shilvock, A.R.; Hoffmann, H.H.; Rosenberg, B.R.; et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell 2018, 172, 811–824.e814. [Google Scholar] [CrossRef] [PubMed]
- de Reuver, R.; Verdonck, S.; Dierick, E.; Nemegeer, J.; Hessmann, E.; Ahmad, S.; Jans, M.; Blancke, G.; Van Nieuwerburgh, F.; Botzki, A.; et al. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 2022, 607, 784–789. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, N.W.; Ames, J.M.; Maurano, M.; Chu, L.H.; Somfleth, K.Y.; Gokhale, N.S.; Werner, M.; Snyder, J.M.; Lichauco, K.; Savan, R.; et al. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 2022, 607, 769–775. [Google Scholar] [CrossRef]
- Ota, H.; Sakurai, M.; Gupta, R.; Valente, L.; Wulff, B.E.; Ariyoshi, K.; Iizasa, H.; Davuluri, R.V.; Nishikura, K. ADAR1 forms a complex with Dicer to promote microRNA processing and RNA-induced gene silencing. Cell 2013, 153, 575–589. [Google Scholar] [CrossRef]
- Hajji, K.; Sedmik, J.; Cherian, A.; Amoruso, D.; Keegan, L.P.; O’Connell, M.A. ADAR2 enzymes: Efficient site-specific RNA editors with gene therapy aspirations. RNA 2022, 28, 1281–1297. [Google Scholar] [CrossRef]
- Higuchi, M.; Maas, S.; Single, F.N.; Hartner, J.; Rozov, A.; Burnashev, N.; Feldmeyer, D.; Sprengel, R.; Seeburg, P.H. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 2000, 406, 78–81. [Google Scholar] [CrossRef]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Oakes, E.; Anderson, A.; Cohen-Gadol, A.; Hundley, H.A. Adenosine Deaminase That Acts on RNA 3 (ADAR3) Binding to Glutamate Receptor Subunit B Pre-mRNA Inhibits RNA Editing in Glioblastoma. J. Biol. Chem. 2017, 292, 4326–4335. [Google Scholar] [CrossRef]
- Raghava Kurup, R.; Oakes, E.K.; Manning, A.C.; Mukherjee, P.; Vadlamani, P.; Hundley, H.A. RNA binding by ADAR3 inhibits adenosine-to-inosine editing and promotes expression of immune response protein MAVS. J. Biol. Chem. 2022, 298, 102267. [Google Scholar] [CrossRef] [PubMed]
- Mladenova, D.; Barry, G.; Konen, L.M.; Pineda, S.S.; Guennewig, B.; Avesson, L.; Zinn, R.; Schonrock, N.; Bitar, M.; Jonkhout, N.; et al. Adar3 Is Involved in Learning and Memory in Mice. Front. Neurosci. 2018, 12, 243. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.E. Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral. Virology 2011, 411, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Gatherer, D.; Depledge, D.P.; Hartley, C.A.; Szpara, M.L.; Vaz, P.K.; Benko, M.; Brandt, C.R.; Bryant, N.A.; Dastjerdi, A.; Doszpoly, A.; et al. ICTV Virus Taxonomy Profile: Herpesviridae 2021. J. Gen. Virol. 2021, 102, 001673. [Google Scholar] [CrossRef]
- Pellett, E.P.; Roizman, B. Herpesviridae. In Fields of Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2017; Volume 2. [Google Scholar]
- Murthy, S.; Couacy-Hymann, E.; Metzger, S.; Nowak, K.; De Nys, H.; Boesch, C.; Wittig, R.; Jarvis, M.A.; Leendertz, F.H.; Ehlers, B. Absence of frequent herpesvirus transmission in a nonhuman primate predator-prey system in the wild. J. Virol. 2013, 87, 10651–10659. [Google Scholar] [CrossRef]
- Rosani, U.; Gaia, M.; Delmont, T.O.; Krupovic, M. Tracing the invertebrate herpesviruses in the global sequence datasets. Front. Mar. Sci. 2023, 10, 1159754. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: New York, NY, USA, 2013; Volume 2. [Google Scholar]
- Cliffe, A.R.; Garber, D.A.; Knipe, D.M. Transcription of the herpes simplex virus latency-associated transcript promotes the formation of facultative heterochromatin on lytic promoters. J. Virol. 2009, 83, 8182–8190. [Google Scholar] [CrossRef]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C.; Ciacci-Zanella, J.; Stone, M.; Henderson, G.; Yukht, A.; Slanina, S.M.; Hofman, F.M.; Ghiasi, H.; Nesburn, A.B.; et al. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science 2000, 287, 1500–1503. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol. 2001, 75, 6660–6675. [Google Scholar] [CrossRef]
- Wang, Q.Y.; Zhou, C.; Johnson, K.E.; Colgrove, R.C.; Coen, D.M.; Knipe, D.M. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc. Natl. Acad. Sci. USA 2005, 102, 16055–16059. [Google Scholar] [CrossRef] [PubMed]
- Jurak, I.; Kramer, M.F.; Mellor, J.C.; van Lint, A.L.; Roth, F.P.; Knipe, D.M.; Coen, D.M. Numerous Conserved and Divergent MicroRNAs Expressed by Herpes Simplex Viruses 1 and 2. J. Virol. 2010, 84, 4659–4672. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Nagel, M.A.; Cohrs, R.J.; Gilden, D.H.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Wang, K.; Tang, S.; Krause, P.R.; Mont, E.K.; Cohen, J.I.; Cullen, B.R. Identification of viral microRNAs expressed in human sacral ganglia latently infected with herpes simplex virus 2. J. Virol. 2010, 84, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.Y.; Liu, X.J.; Chen, X.Q.; Zhou, X.S.; Du, T.; Roizman, B.; Zhou, G.Y. miR-H28 and miR-H29 expressed late in productive infection are exported and restrict HSV-1 replication and spread in recipient cells. Proc. Natl. Acad. Sci. USA 2016, 113, E894–E901. [Google Scholar] [CrossRef]
- Cokaric Brdovcak, M.; Zubkovic, A.; Ferencic, A.; Sosa, I.; Stemberga, V.; Cuculic, D.; Rokic, F.; Vugrek, O.; Hackenberg, M.; Jurak, I. Herpes simplex virus 1 miRNA sequence variations in latently infected human trigeminal ganglia. Virus Res. 2018, 256, 90–95. [Google Scholar] [CrossRef]
- Pan, D.; Pesola, J.M.; Li, G.; McCarron, S.; Coen, D.M. Mutations Inactivating Herpes Simplex Virus 1 MicroRNA miR-H2 Do Not Detectably Increase ICP0 Gene Expression in Infected Cultured Cells or Mouse Trigeminal Ganglia. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Flores, O.; Nakayama, S.; Whisnant, A.W.; Javanbakht, H.; Cullen, B.R.; Bloom, D.C. Mutational inactivation of herpes simplex virus 1 microRNAs identifies viral mRNA targets and reveals phenotypic effects in culture. J. Virol. 2013, 87, 6589–6603. [Google Scholar] [CrossRef]
- Barrozo, E.R.; Nakayama, S.; Singh, P.; Neumann, D.M.; Bloom, D.C. Herpes Simplex Virus 1 MicroRNA miR-H8 Is Dispensable for Latency and Reactivation In Vivo. J. Virol. 2021, 95, e02179-20. [Google Scholar] [CrossRef]
- Jurak, I.; Silverstein, L.B.; Sharma, M.; Coen, D.M. Herpes simplex virus is equipped with RNA- and protein-based mechanisms to repress expression of ATRX, an effector of intrinsic immunity. J. Virol. 2012, 86, 10093–10102. [Google Scholar] [CrossRef] [PubMed]
- Zubkovic, A.; Gomes, C.; Parchure, A.; Cesarec, M.; Ferencic, A.; Rokic, F.; Jakovac, H.; Whitford, A.L.; Dochnal, S.A.; Cliffe, A.R.; et al. HSV-1 miRNAs are post-transcriptionally edited in latently infected human ganglia. J. Virol. 2023, e0073023. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.; Gilden, D. Varicela-Zoster Virus. In Fields of Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: New York, NY, USA, 2013; Volume 2. [Google Scholar]
- Depledge, D.P.; Sadaoka, T.; Ouwendijk, W.J.D. Molecular Aspects of Varicella-Zoster Virus Latency. Viruses 2018, 10, 349. [Google Scholar] [CrossRef] [PubMed]
- Prazsak, I.; Moldovan, N.; Balazs, Z.; Tombacz, D.; Megyeri, K.; Szucs, A.; Csabai, Z.; Boldogkoi, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genom. 2018, 19, 873. [Google Scholar] [CrossRef]
- Osterrieder, N.; Wallaschek, N.; Kaufer, B.B. Herpesvirus Genome Integration into Telomeric Repeats of Host Cell Chromosomes. Annu. Rev. Virol. 2014, 1, 215–235. [Google Scholar] [CrossRef]
- Osterrieder, N.; Kamil, J.P.; Schumacher, D.; Tischer, B.K.; Trapp, S. Marek’s disease virus: From miasma to model. Nat. Rev. Microbiol. 2006, 4, 283–294. [Google Scholar] [CrossRef]
- Cantello, J.L.; Anderson, A.S.; Morgan, R.W. Identification of latency-associated transcripts that map antisense to the ICP4 homolog gene of Marek’s disease virus. J. Virol. 1994, 68, 6280–6290. [Google Scholar] [CrossRef]
- Figueroa, T.; Boumart, I.; Coupeau, D.; Rasschaert, D. Hyperediting by ADAR1 of a new herpesvirus lncRNA during the lytic phase of the oncogenic Marek’s disease virus. J. Gen. Virol. 2016, 97, 2973–2988. [Google Scholar] [CrossRef]
- Mocarski, E.S.; Shenk, T.; Griffiths, P.D.; Pass, R.F. Cytomegaloviruses. In Fields of Virology, 6th ed.; Lippincott Williams & Wilklins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Goodrum, F. The complex biology of human cytomegalovirus latency. Adv. Virus Res. 2022, 112, 31–85. [Google Scholar] [CrossRef]
- Slavuljica, I.; Krmpotic, A.; Jonjic, S. Manipulation of NKG2D ligands by cytomegaloviruses: Impact on innate and adaptive immune response. Front. Immunol. 2011, 2, 85. [Google Scholar] [CrossRef]
- Nachmani, D.; Lankry, D.; Wolf, D.G.; Mandelboim, O. The human cytomegalovirus microRNA miR-UL112 acts synergistically with a cellular microRNA to escape immune elimination. Nat. Immunol. 2010, 11, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Dolken, L.; Perot, J.; Cognat, V.; Alioua, A.; John, M.; Soutschek, J.; Ruzsics, Z.; Koszinowski, U.; Voinnet, O.; Pfeffer, S. Mouse cytomegalovirus microRNAs dominate the cellular small RNA profile during lytic infection and show features of posttranscriptional regulation. J. Virol. 2007, 81, 13771–13782. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-Barr Virus. In Fields of Virology, 6th ed.; Lippincott Williams & Wilklins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Frappier, L. Epstein-Barr virus: Current questions and challenges. Tumour Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef] [PubMed]
- Bjornevik, K.; Cortese, M.; Healy, B.C.; Kuhle, J.; Mina, M.J.; Leng, Y.; Elledge, S.J.; Niebuhr, D.W.; Scher, A.I.; Munger, K.L.; et al. Longitudinal analysis reveals high prevalence of Epstein-Barr virus associated with multiple sclerosis. Science 2022, 375, 296–301. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Cullen, B.R. EBV Noncoding RNAs. Curr. Top. Microbiol. Immunol. 2015, 391, 181–217. [Google Scholar] [CrossRef]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editing of Epstein-Barr Virus-encoded BART6 MicroRNAs Controls Their Dicer Targeting and Consequently Affects Viral Latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar] [CrossRef]
- Lei, T.; Yuen, K.S.; Tsao, S.W.; Chen, H.; Kok, K.H.; Jin, D.Y. Perturbation of biogenesis and targeting of Epstein-Barr virus-encoded miR-BART3 microRNA by adenosine-to-inosine editing. J. Gen. Virol. 2013, 94, 2739–2744. [Google Scholar] [CrossRef]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef]
- Skalsky, R.L.; Corcoran, D.L.; Gottwein, E.; Frank, C.L.; Kang, D.; Hafner, M.; Nusbaum, J.D.; Feederle, R.; Delecluse, H.J.; Luftig, M.A.; et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012, 8, e1002484. [Google Scholar] [CrossRef]
- Cao, S.; Moss, W.; O’Grady, T.; Concha, M.; Strong, M.J.; Wang, X.; Yu, Y.; Baddoo, M.; Zhang, K.; Fewell, C.; et al. New Noncoding Lytic Transcripts Derived from the Epstein-Barr Virus Latency Origin of Replication, oriP, Are Hyperedited, Bind the Paraspeckle Protein, NONO/p54nrb, and Support Viral Lytic Transcription. J. Virol. 2015, 89, 7120–7132. [Google Scholar] [CrossRef]
- Damania, B.A.; Ceserman, E. Kaposi’s Sarcoma-Associated Herpesvirus. In Fields of Virology, 6th ed.; Lippincott Williams & Wilklins: Philadelphia, PA, USA, 2013; Volume 2. [Google Scholar]
- Dissinger, N.J.; Damania, B. Recent advances in understanding Kaposi’s sarcoma-associated herpesvirus. F1000Research 2016, 5, 740. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Raja, A.N.; Shamulailatpam, P.; Manzano, M.; Schipma, M.J.; Casey, J.L.; Gottwein, E. MicroRNA-mediated transformation by the Kaposi’s sarcoma-associated herpesvirus Kaposin locus. J. Virol. 2015, 89, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of microRNAs of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef]
- Gottwein, E.; Cai, X.; Cullen, B.R. A novel assay for viral microRNA function identifies a single nucleotide polymorphism that affects Drosha processing. J. Virol. 2006, 80, 5321–5326. [Google Scholar] [CrossRef]
- Gandy, S.Z.; Linnstaedt, S.D.; Muralidhar, S.; Cashman, K.A.; Rosenthal, L.J.; Casey, J.L. RNA editing of the human herpesvirus 8 kaposin transcript eliminates its transforming activity and is induced during lytic replication. J. Virol. 2007, 81, 13544–13551. [Google Scholar] [CrossRef]
- Rajendren, S.; Ye, X.; Dunker, W.; Richardson, A.; Karijolich, J. The cellular and KSHV A-to-I RNA editome in primary effusion lymphoma and its role in the viral lifecycle. Nat. Commun. 2023, 14, 1367. [Google Scholar] [CrossRef]
- Liu, Q.; Rao, Y.; Tian, M.; Zhang, S.; Feng, P. Modulation of Innate Immune Signaling Pathways by Herpesviruses. Viruses 2019, 11, 572. [Google Scholar] [CrossRef]
- O’Connor, C.M.; Sen, G.C. Innate Immune Responses to Herpesvirus Infection. Cells 2021, 10, 2122. [Google Scholar] [CrossRef]
- Zhang, H.; Ni, G.; Damania, B. ADAR1 Facilitates KSHV Lytic Reactivation by Modulating the RLR-Dependent Signaling Pathway. Cell Rep. 2020, 31, 107564. [Google Scholar] [CrossRef] [PubMed]
- Mushegian, A.; Karin, E.L.; Pupko, T. Sequence analysis of malacoherpesvirus proteins: Pan-herpesvirus capsid module and replication enzymes with an ancient connection to “Megavirales”. Virology 2018, 513, 114–128. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Bai, C.M.; Maso, L.; Shapiro, M.; Abbadi, M.; Domeneghetti, S.; Wang, C.M.; Cendron, L.; MacCarthy, T.; Venier, P. A-to-I editing of Malacoherpesviridae RNAs supports the antiviral role of ADAR1 in mollusks. BMC Evol. Biol. 2019, 19, 149. [Google Scholar] [CrossRef] [PubMed]
- Rosani, U.; Bortoletto, E.; Montagnani, C.; Venier, P. ADAR-Editing during Ostreid Herpesvirus 1 Infection in Crassostrea gigas: Facts and Limitations. mSphere 2022, 7, e0001122. [Google Scholar] [CrossRef]
- Bai, C.M.; Rosani, U.; Zhang, X.; Xin, L.S.; Bortoletto, E.; Wegner, K.M.; Wang, C.M. Viral Decoys: The Only Two Herpesviruses Infecting Invertebrates Evolved Different Transcriptional Strategies to Deflect Post-Transcriptional Editing. Viruses 2021, 13, 1971. [Google Scholar] [CrossRef]
- Cheng, A.Z.; Moraes, S.N.; Shaban, N.M.; Fanunza, E.; Bierle, C.J.; Southern, P.J.; Bresnahan, W.A.; Rice, S.A.; Harris, R.S. APOBECs and Herpesviruses. Viruses 2021, 13, 390. [Google Scholar] [CrossRef]
- Shen, H.Q.; An, O.; Ren, X.; Song, Y.Y.; Tang, Z.J.; Ke, X.Y.; Han, J.; Tay, D.J.T.; Ng, V.H.E.; Molias, F.B.; et al. ADARs act as potent regulators of circular transcriptome in cancer. Nat. Commun. 2022, 13, 1508. [Google Scholar] [CrossRef]
- Kapoor, U.; Licht, K.; Amman, F.; Jakobi, T.; Martin, D.; Dieterich, C.; Jantsch, M.F. ADAR-deficiency perturbs the global splicing landscape in mouse tissues. Genome Res. 2020, 30, 1107–1118. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in human diseases and potential use as biomarkers. Cell Death Dis. 2021, 12, 468. [Google Scholar] [CrossRef]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Li, S.; Yang, N.; Zou, Y.; Zheng, D.; Xiao, T. Recent progress in circular RNAs in human cancers. Cancer Lett. 2017, 404, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Oh, D.; Dremel, S.; Mahesh, G.; Koparde, V.N.; Duncan, G.; Andresson, T.; Ziegelbauer, J.M. A virus-induced circular RNA maintains latent infection of Kaposi’s sarcoma herpesvirus. Proc. Natl. Acad. Sci. USA 2023, 120, e2212864120. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef]
- Lei, M.; Liu, Y.; Samuel, C.E. Adenovirus VAI RNA antagonizes the RNA-editing activity of the ADAR adenosine deaminase. Virology 1998, 245, 188–196. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Aublin-Gex, A.; Ruggieri, A.; Meyniel-Schicklin, L.; Pradezynski, F.; Davoust, N.; Chantier, T.; Tafforeau, L.; Mangeot, P.E.; Ciancia, C.; et al. The interactomes of influenza virus NS1 and NS2 proteins identify new host factors and provide insights for ADAR1 playing a supportive role in virus replication. PLoS Pathog. 2013, 9, e1003440. [Google Scholar] [CrossRef]
- Liu, Y.; Wolff, K.C.; Jacobs, B.L.; Samuel, C.E. Vaccinia virus E3L interferon resistance protein inhibits the interferon-induced adenosine deaminase A-to-I editing activity. Virology 2001, 289, 378–387. [Google Scholar] [CrossRef]


{kind=link}
| Virus Taxonomy | Virus | ADAR Activity | Ref. | ||
|---|---|---|---|---|---|
| Herpesvirales | Ortoherpesviridae | Alphaherpesvirinae | HSV-1 (HHV-1) | ADAR1 expression levels maintained during productive infection. Editing of HSV-1 miR-H2-3p in latency and to lesser extent in productive infection. Function: increased targeting repertoire of miR-H2-3p. | [13,49,54] |
| VZV (HHV-3) | Dynamics of ADAR expression levels: unknown. Editing of lncRNA NTO3 (antisense to ORF63). Function: unknown. | [57] | |||
| GaHV-2 | Dynamics of ADAR expression levels: unknown. Editing of ERL lncRNA. Function: unknown. | [61] | |||
| Betaherpesvirinae | HCMV (HHV-5) | ADAR1 p110 is upregulated in productive infection. Editing of host miR-376a. Function: edited miRNA gains specificity to downregulates HLA-E and abolishes targeting of MICB (ligand of activating NKG2D receptor), facilitating elimination of HCMV infected cells. | [13] | ||
| Gammaherpesvirinae | EBV (HHV-4) | Dynamics of ADAR expression levels: unknown. Editing of pri-BHRF1-1, pri-miR-BART3,-BART6, -BART8, -BART11, and -BART16. Editing of vlncRNA oriPtL and oriPtR. Functions: affected Drosha processing of pri-miR-BART6 and -BART3 resulting in lower levels of miRNAs, and loss of posttranscriptional regulation of their targets (Dicer). miR-BART3 seed sequence editing abolished Dicer targeting. Functions of edited oriPtL and oriPtR: unknown. | [71,72,75] | ||
| KSHV (HHV-8) | ADAR1 expression levels maintained from latent to lytic infection. * ADAR1 (all forms) increased during reactivation. * Editing of K12 transcript, LANA, RTA, etc., and pri-miR-K12-10, pri-miR-K12-4 Functions: Editing eliminates K12 transforming activity and reduces pri-miR-K12-4 processing by Drosha. Increased repertoire of miR-K12-4 targets. ADAR1 prevents activation of RIG-I signaling and enables efficient virus reactivation. | [14,81,83,84,87] | |||
| Malacoherpesviridae | OsHV-1 HaHV-1 | ADAR1 upregulated in productively infected host. Editing: increased global editing of viral and host transcripts during infection. Function: unknown. | [89,90,91] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanišević, V.; Žilić, L.; Čunko, M.; Fadiga, H.; Munitić, I.; Jurak, I. RNA Editing-Dependent and -Independent Roles of Adenosine Deaminases Acting on RNA Proteins in Herpesvirus Infection—Hints on Another Layer of Complexity. Viruses 2023, 15, 2007. https://doi.org/10.3390/v15102007
Ivanišević V, Žilić L, Čunko M, Fadiga H, Munitić I, Jurak I. RNA Editing-Dependent and -Independent Roles of Adenosine Deaminases Acting on RNA Proteins in Herpesvirus Infection—Hints on Another Layer of Complexity. Viruses. 2023; 15(10):2007. https://doi.org/10.3390/v15102007
Chicago/Turabian StyleIvanišević, Vlatka, Lidia Žilić, Marina Čunko, Hana Fadiga, Ivana Munitić, and Igor Jurak. 2023. "RNA Editing-Dependent and -Independent Roles of Adenosine Deaminases Acting on RNA Proteins in Herpesvirus Infection—Hints on Another Layer of Complexity" Viruses 15, no. 10: 2007. https://doi.org/10.3390/v15102007