
Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Detection of EV RNA
2.2. Gene Amplification and DNA Sequencing
2.3. Phylogenetic Analysis and Evolutionary Dynamics
2.4. Recombination Analysis
2.5. Nucleotide Sequence Accession Numbers
3. Results
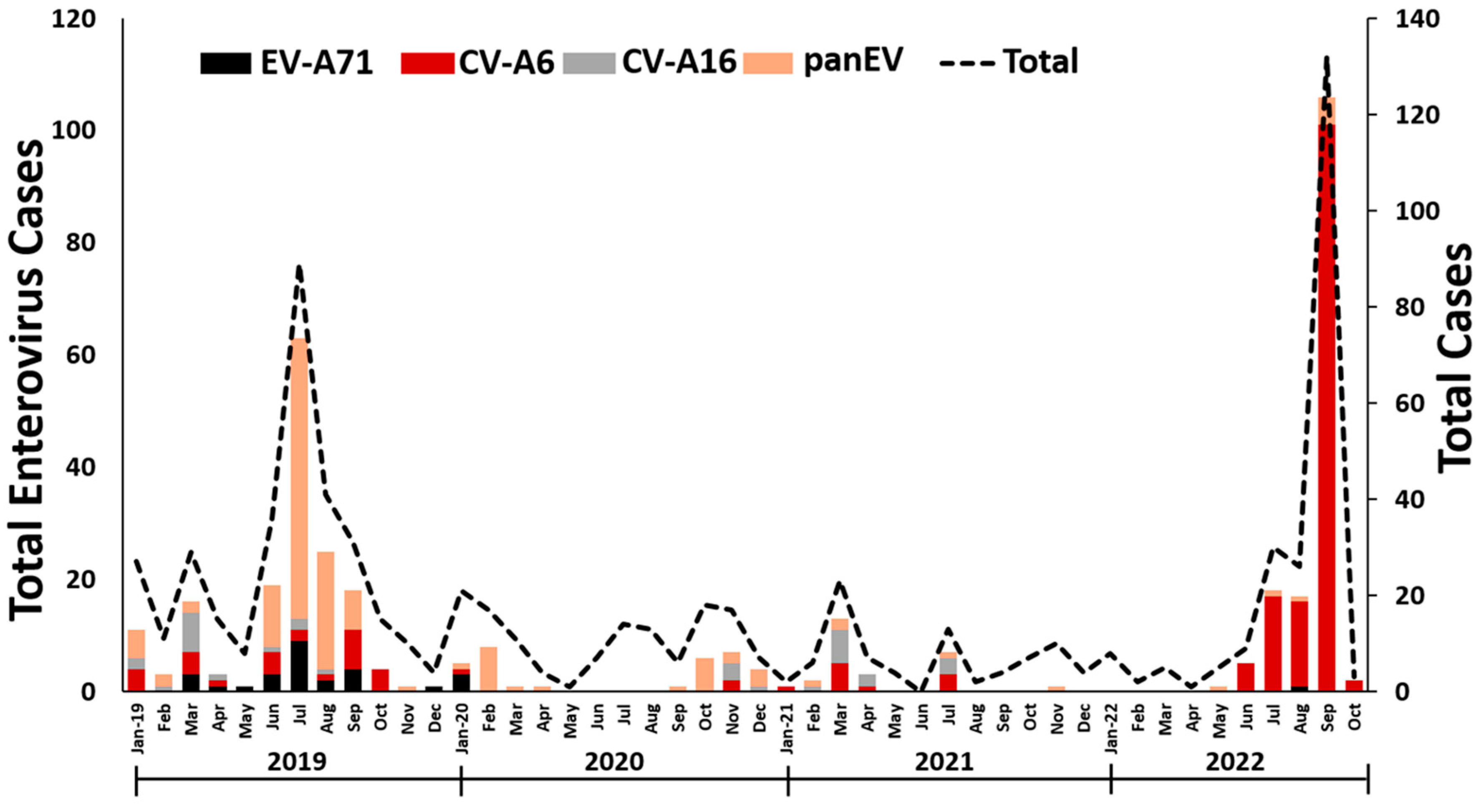
3.1. EV detection in Thailand
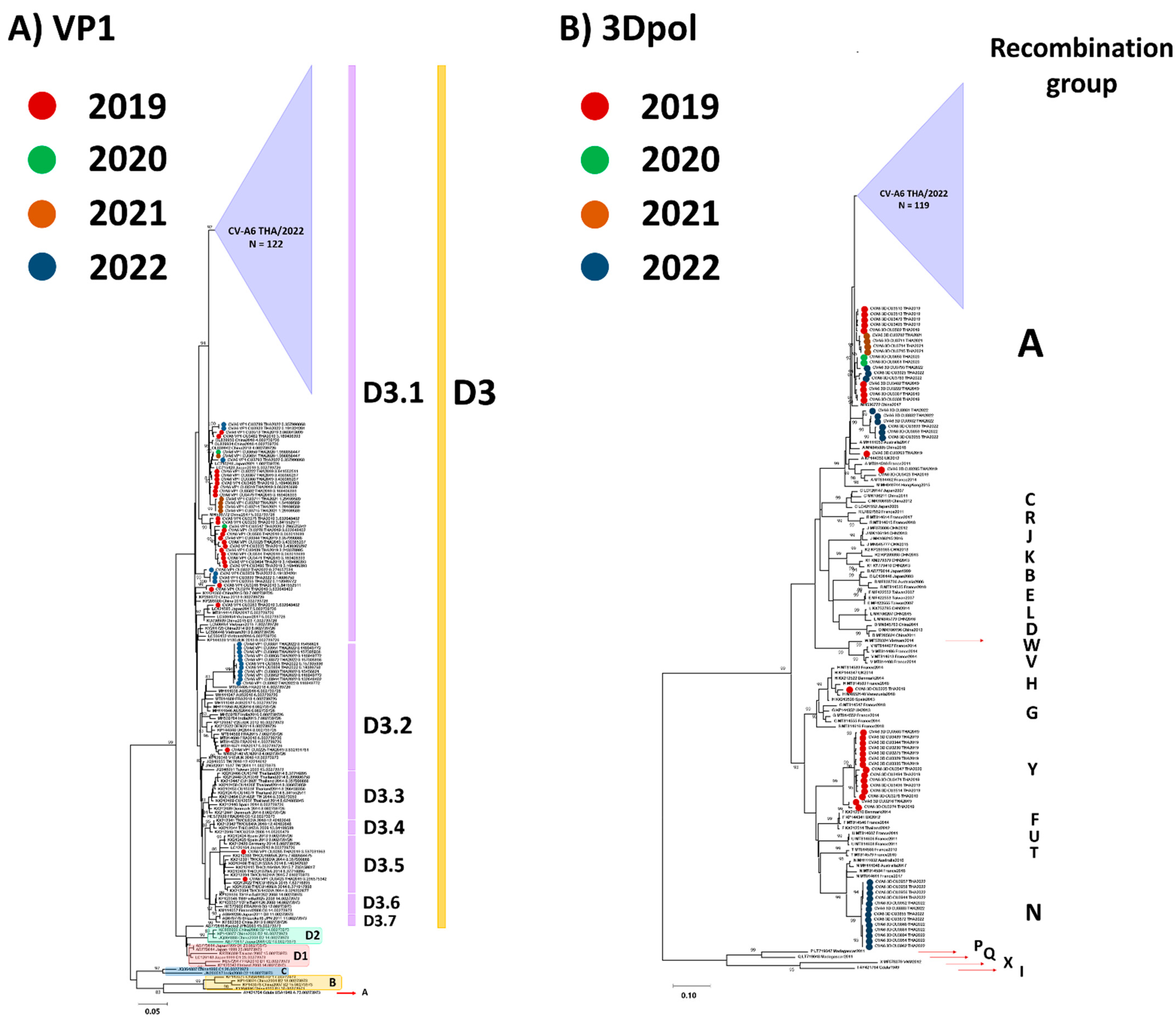
3.2. Phylogenetic Analysis
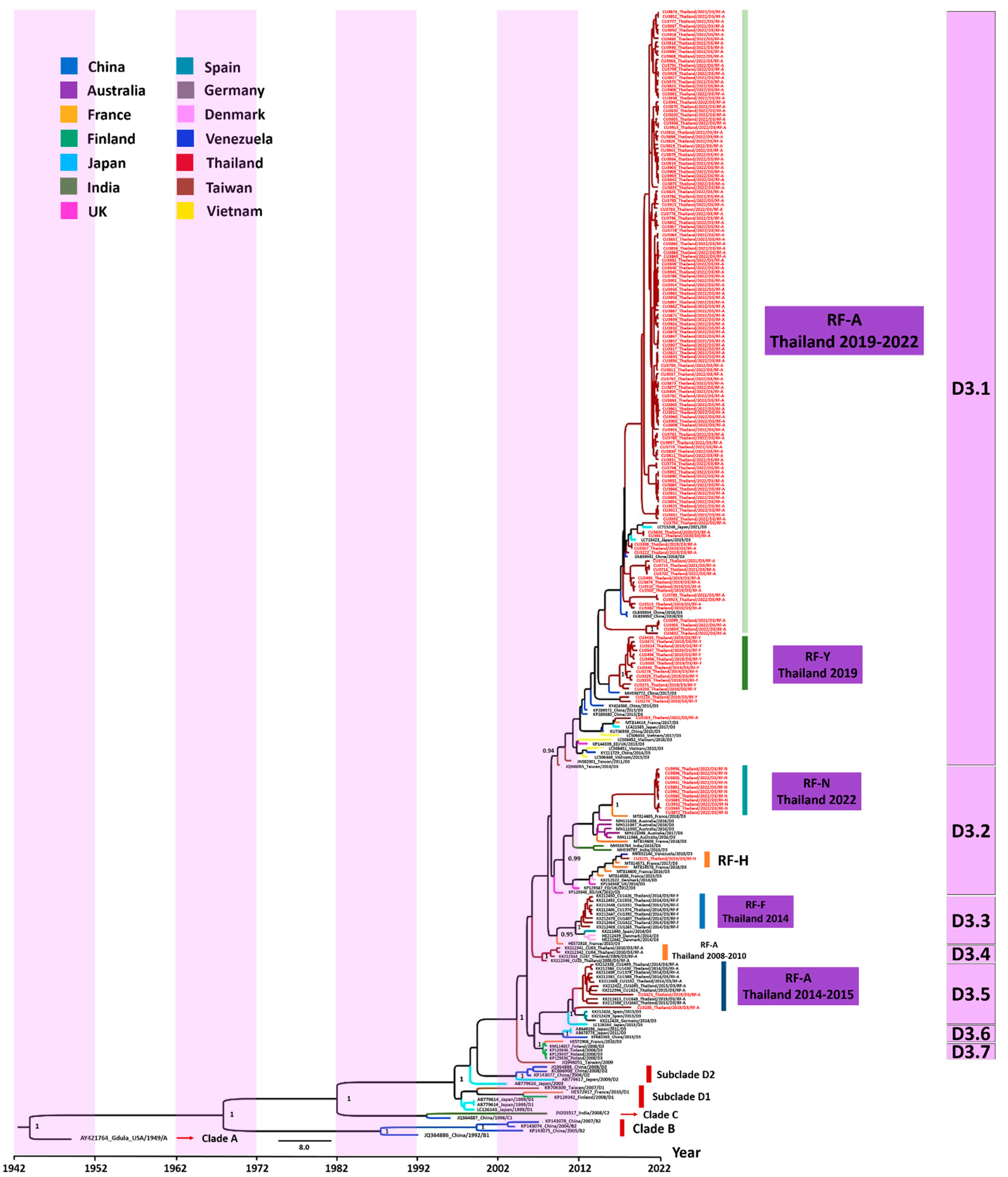
3.3. Evaluation of the Evolutionary History of CV-A6 Using the VP1 Sequence Dataset
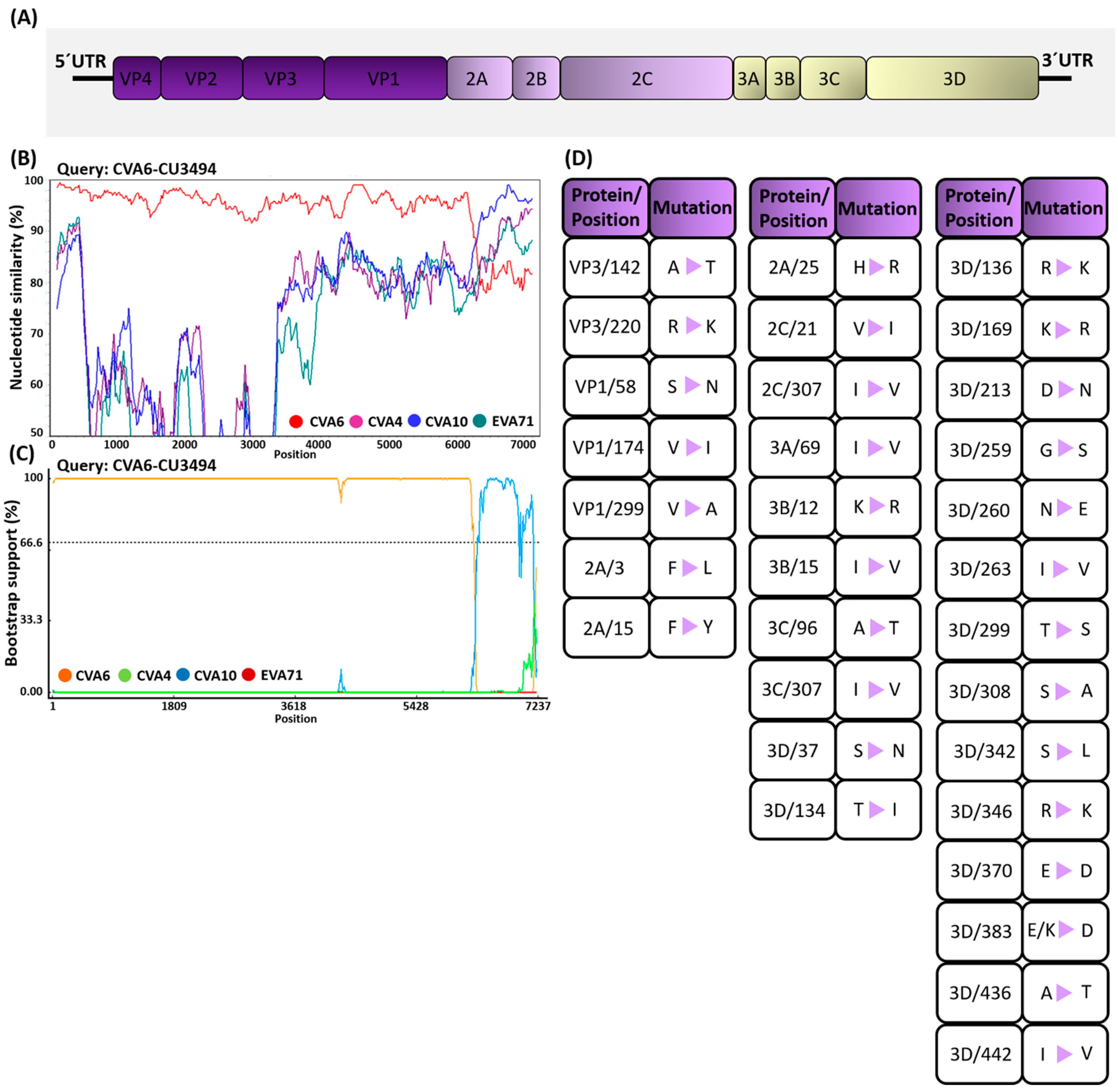
3.4. Analysis of Mutation Profiles in CV-A6 Recombinant Subclade D3/Y Genomes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Osterback, R.; Vuorinen, T.; Linna, M.; Susi, P.; Hyypiä, T.; Waris, M. Coxsackievirus A6 and hand, foot, and mouth disease, Finland. Emerg. Infect. Dis. 2009, 15, 1485–1488. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, E.; Harvala, H.; Österback, R.; Sreenu, V.B.; Thomson, E.; Waris, M.; Simmonds, P. Genetic characterization of human coxsackievirus A6 variants associated with atypical hand, foot and mouth disease: A potential role of recombination in emergence and pathogenicity. J. Gen. Virol. 2015, 96, 1067–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flett, K.; Youngster, I.; Huang, J.; McAdam, A.; Sandora, T.J.; Rennick, M.; Smole, S.; Rogers, S.L.; Nix, W.A.; Oberste, M.S.; et al. Hand, foot, and mouth disease caused by coxsackievirus a6. Emerg. Infect. Dis. 2012, 18, 1702–1704. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Zhang, Y.; Ji, T.; Gu, X.; Yang, Q.; Zhu, S.; Xu, W.; Xu, Y.; Shi, Y.; Huang, X.; et al. Persistent circulation of Coxsackievirus A6 of genotype D3 in mainland of China between 2008 and 2015. Sci. Rep. 2017, 7, 5491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cabrerizo, M.; Tarragó, D.; Muñoz-Almagro, C.; Del Amo, E.; Domínguez-Gil, M.; Eiros, J.M.; López-Miragaya, I.; Pérez, C.; Reina, J.; Otero, A.; et al. Molecular epidemiology of enterovirus 71, coxsackievirus A16 and A6 associated with hand, foot and mouth disease in Spain. Clin. Microbiol. Infect. 2014, 20, O150–O156. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, M.C.; Sarmiento, L.; Resik, S.; Martínez, Y.; Hung, L.H.; Morier, L.; Piñón, A.; Valdéz, O.; Kourí, V.; González, G. Coxsackievirus A6 and enterovirus 71 causing hand, foot and mouth disease in Cuba, 2011–2013. Arch. Virol. 2014, 159, 2451–2455. [Google Scholar] [CrossRef]
- Carmona, R.C.C.; Machado, B.C.; Reis, F.C.; Jorge, A.M.V.; Cilli, A.; Dias, A.M.N.; Morais, D.R.; Leme, L.; Yu, A.L.F.; Silva, M.R.; et al. Hand, foot, and mouth disease outbreak by Coxsackievirus A6 during COVID-19 pandemic in 2021, São Paulo, Brazil. J. Clin. Virol. 2022, 154, 105245. [Google Scholar] [CrossRef]
- Zhang, M.; Chen, X.; Wang, W.; Li, Q.; Xie, Z. Genetic characteristics of Coxsackievirus A6 from children with hand, foot and mouth disease in Beijing, China, 2017–2019. Infect. Genet. Evol. 2022, 106, 105378. [Google Scholar] [CrossRef]
- Mirand, A.; Cohen, R.; Bisseux, M.; Tomba, S.; Sellem, F.C.; Gelbert, N.; Béchet, S.; Frandji, B.; Archimbaud, C.; Brebion, A.; et al. A large-scale outbreak of hand, foot and mouth disease, France, as at 28 September 2021. Euro Surveill. 2021, 26, 2100978. [Google Scholar] [CrossRef]
- Lau, S.K.P.; Zhao, P.S.H.; Sridhar, S.; Yip, C.C.Y.; Aw-Yong, K.L.; Chow, E.Y.Y.; Cheung, K.C.M.; Hui, R.W.H.; Leung, R.Y.H.; Lai, Y.S.K.; et al. Molecular epidemiology of coxsackievirus A6 circulating in Hong Kong reveals common neurological manifestations and emergence of novel recombinant groups. J. Clin. Virol. 2018, 108, 43–49. [Google Scholar] [CrossRef]
- Saxena, V.K.; Pawar, S.D.; Qureshi, T.H.I.H.; Surve, P.; Yadav, P.; Nabi, F.; Mendadkar, R. Isolation and molecular characterization of coxsackievirus A6 and coxsackievirus A16 from a case of recurrent Hand, Foot and Mouth Disease in Mumbai, Maharashtra, India, 2018. Virusdisease 2020, 31, 56–60. [Google Scholar] [CrossRef]
- Mizuta, K.; Tanaka, S.; Komabayashi, K.; Aoki, Y.; Itagaki, T.; Katsushima, F.; Katsushima, Y.; Yoshida, H.; Ito, S.; Matsuzaki, Y.; et al. Phylogenetic and antigenic analyses of coxsackievirus A6 isolates in Yamagata, Japan between 2001 and 2017. Vaccine 2019, 37, 1109–1117. [Google Scholar] [CrossRef]
- Ceylan, A.N.; Turel, O.; Gultepe, B.S.; Inan, E.; Turkmen, A.V.; Doymaz, M.Z. Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6: A Preliminary Report from Istanbul. Pol. J. Microbiol. 2019, 68, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Lizasoain, A.; Piegas, S.; Victoria, M.; Da Silva, E.E.; Colina, R. Hand-foot-and-mouth disease in uruguay: Coxsackievirus A6 identified as causative of an outbreak in a rural childcare center. J. Med. Virol. 2020, 92, 167–173. [Google Scholar] [CrossRef]
- Hoa-Tran, T.N.; Dao, A.T.H.; Nguyen, A.T.; Kataoka, C.; Takemura, T.; Pham, C.H.; Vu, H.M.; Hong, T.T.T.; Ha, N.T.V.; Duong, T.N.; et al. Coxsackieviruses A6 and A16 associated with hand, foot, and mouth disease in Vietnam, 2008–2017: Essential information for rational vaccine design. Vaccine 2020, 38, 8273–8285. [Google Scholar] [CrossRef]
- Cisterna, D.M.; Lema, C.L.; Martinez, L.M.; Verón, E.; Contarino, L.P.; Acosta, D.; Freire, M.C. Atypical hand, foot, and mouth disease caused by Coxsackievirus A6 in Argentina in 2015. Rev. Argent. Microbiol. 2019, 51, 140–143. [Google Scholar] [CrossRef]
- Justino, M.C.A.; da S Mesquita, D.; Souza, M.F.; Farias, F.P.; Dos S Alves, J.C.; Ferreira, J.L.; Lopes, D.P.; Tavares, F.N. Atypical hand-foot-mouth disease in Belém, Amazon region, northern Brazil, with detection of coxsackievirus A6. J. Clin. Virol. 2020, 126, 104307. [Google Scholar] [CrossRef]
- Gao, Y.; Ma, G.; Xiao, Y.; Cai, Q.; Chen, Y.; Shi, P.; Wang, K.; Shen, Y.; Shi, C. An Outbreak of Coxsackievirus A6 Infection in Adults of a Collective Unit, China, 2019. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 6607294. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Marva, E.; Weil, M.; Shulman, L.M.; Gencylmaz, N.; Sheffer, S.; Wolf, D.G.; Molho-Pessach, V. Coxsackievirus A6 Polymorphic Exanthem in Israeli Children. Acta Derm. Venereol. 2016, 96, 546–549. [Google Scholar] [CrossRef] [Green Version]
- Broccolo, F.; Drago, F.; Ciccarese, G.; Genoni, A.; Puggioni, A.; Rosa, G.M.; Parodi, A.; Manukyan, H.; Laassri, M.; Chumakov, K.; et al. Severe atypical hand-foot-and-mouth disease in adults due to coxsackievirus A6: Clinical presentation and phylogenesis of CV-A6 strains. J. Clin. Virol. 2019, 110, 1–6. [Google Scholar] [CrossRef]
- No, T.H.; Jo, K.M.; Jung, S.Y.; Kim, M.R.; Kim, J.Y.; Park, C.S.; Kym, S. Coxsackievirus A6-induced Hand-Foot-and-Mouth Disease Mimicking Stevens-Johnson Syndrome in an Immunocompetent Adult. Infect. Chemother. 2020, 52, 634–640. [Google Scholar] [CrossRef] [PubMed]
- Puenpa, J.; Wanlapakorn, N.; Vongpunsawad, S.; Poovorawan, Y. The History of Enterovirus A71 Outbreaks and Molecular Epidemiology in the Asia-Pacific Region. J. Biomed. Sci. 2019, 26, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Liu, F.; Ren, M.; Nie, T.; Cui, J.; Chang, Z.; Li, Z. Basic Reproduction Number of Enterovirus 71 and Coxsackievirus A16 and A6: Evidence From Outbreaks of Hand, Foot, and Mouth Disease in China Between 2011 and 2018. Clin. Infect. Dis. 2021, 73, e2552–e2559. [Google Scholar] [CrossRef] [PubMed]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Ooi, M.H.; Wong, S.C.; Podin, Y.; Akin, W.; del Sel, S.; Mohan, A.; Chieng, C.H.; Perera, D.; Clear, D.; Wong, D.; et al. Human enterovirus 71 disease in Sarawak, Malaysia: A prospective clinical, virological, and molecular epidemiological study. Clin. Infect. Dis. 2007, 44, 646–656. [Google Scholar] [CrossRef]
- Wu, W.H.; Kuo, T.C.; Lin, Y.T.; Huang, S.W.; Liu, H.F.; Wang, J.; Chen, Y.M. Molecular epidemiology of enterovirus 71 infection in the central region of Taiwan from 2002 to 2012. PLoS ONE 2013, 8, e83711. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.J.; Hyeon, J.Y.; Hwang, S.; Lee, Y.P.; Lee, S.W.; Yoo, J.S.; Kang, B.; Ahn, J.B.; Jeong, Y.S.; Lee, J.W. Epidemiology and virologic investigation of human enterovirus 71 infection in the Republic of Korea from 2007 to 2012: A nationwide cross-sectional study. BMC Infect. Dis. 2016, 16, 425. [Google Scholar] [CrossRef] [Green Version]
- Jiang, P.; Liu, Y.; Ma, H.C.; Paul, A.V.; Wimmer, E. Picornavirus morphogenesis. Microbiol. Mol. Biol. Rev. 2014, 78, 418–437. [Google Scholar] [CrossRef] [Green Version]
- Santti, J.; Hyypiä, T.; Kinnunen, L.; Salminen, M. Evidence of recombination among enteroviruses. J. Virol. 1999, 73, 8741–8749. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef] [Green Version]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating Human enterovirus B: Independent evolution of structural and non-structural genome regions. J. Gen. Virol. 2005, 86, 3281–3290. [Google Scholar] [CrossRef]
- Li, C.; Qiao, Q.; Hao, S.B.; Dong, Z.; Zhao, L.; Ji, J.; Wang, Z.Y.; Wen, H.L. Nonstructural Protein 2A Modulates Replication and Virulence of Enterovirus. Virus Res. 2018, 244, 262–269. [Google Scholar] [CrossRef]
- Puenpa, J.; Suwannakarn, K.; Chansaenroj, J.; Vongpunsawad, S.; Poovorawan, Y. Development of single-step multiplex real-time RT-PCR assays for rapid diagnosis of enterovirus 71, coxsackievirus A6, and A16 in patients with hand, foot, and mouth disease. J. Virol. Methods 2017, 248, 92–99. [Google Scholar] [CrossRef]
- Nix, W.A.; Oberste, M.S.; Pallansch, M.A. Sensitive, seminested PCR amplification of VP1 sequences for direct identification of all enterovirus serotypes from original clinical specimens. J. Clin. Microbiol. 2006, 44, 2698–2704. [Google Scholar] [CrossRef] [Green Version]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids. Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef] [Green Version]
- Bouckaert, R.; Vaughan, T.G.; Barido-Sottani, J.; Duchêne, S.; Fourment, M.; Gavryushkina, A.; Heled, J.; Jones, G.; Kühnert, D.; De Maio, N.; et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019, 15, e1006650. [Google Scholar] [CrossRef] [Green Version]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Zhang, Y.; Han, Z.; Xu, W.; Xiao, J.; Wang, X.; Wang, J.; Yang, J.; Yu, Q.; Yu, D.; et al. Genetic recombination in fast-spreading coxsackievirus A6 variants: A potential role in evolution and pathogenicity. Virus Evol. 2020, 6, veaa048. [Google Scholar] [CrossRef]
- Tomba Ngangas, S.; Bisseux, M.; Jugie, G.; Lambert, C.; Cohen, R.; Werner, A.; Archimbaud, C.; Henquell, C.; Mirand, A.; Bailly, J.L. Coxsackievirus A6 Recombinant Subclades D3/A and D3/H Were Predominant in Hand-Foot-And-Mouth Disease Outbreaks in the Paediatric Population, France, 2010–2018. Viruses 2022, 14, 1078. [Google Scholar] [CrossRef]
- Puenpa, J.; Vongpunsawad, S.; Österback, R.; Waris, M.; Eriksson, E.; Albert, J.; Midgley, S.; Fischer, T.K.; Eis-Hübinger, A.M.; Cabrerizo, M.; et al. Molecular epidemiology and the evolution of human coxsackievirus A6. J. Gen. Virol. 2016, 97, 3225–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di, B.; Zhang, Y.; Xie, H.; Li, X.; Chen, C.; Ding, P.; He, P.; Wang, D.; Geng, J.; Luo, L.; et al. Circulation of Coxsackievirus A6 in hand-foot-mouth disease in Guangzhou, 2010–2012. Virol. J. 2014, 11, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukashev, A.N.; Lashkevich, V.A.; Ivanova, O.E.; Koroleva, G.A.; Hinkkanen, A.E.; Ilonen, J. Recombination in circulating enteroviruses. J. Virol. 2003, 77, 10423–10431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ang, P.Y.; Chong, C.W.H.; Alonso, S. Viral determinants that drive Enterovirus-A71 fitness and virulence. Emerg. Microbes. Infect. 2021, 10, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Puenpa, J.; Chieochansin, T.; Linsuwanon, P.; Korkong, S.; Thongkomplew, S.; Vichaiwattana, P.; Theamboonlers, A.; Poovorawan, Y. Hand, foot, and mouth disease caused by coxsackievirus A6, Thailand, 2012. Emerg. Infect. Dis. 2013, 19, 641–643. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.P.; Goh, K.T.; Chong, C.Y.; Teo, E.S.; Lau, G.; Ling, A.E. Epidemic hand, foot and mouth disease caused by human enterovirus 71, Singapore. Emerg. Infect. Dis. 2003, 9, 78–85. [Google Scholar] [CrossRef]
- Linsuwanon, P.; Puenpa, J.; Huang, S.W.; Wang, Y.F.; Mauleekoonphairoj, J.; Wang, J.R.; Poovorawan, Y. Epidemiology and seroepidemiology of human enterovirus 71 among Thai populations. J. Biomed. Sci. 2014, 21, 16. [Google Scholar] [CrossRef] [Green Version]
- Feder, H.M., Jr.; Bennett, N.; Modlin, J.F. Atypical hand, foot, and mouth disease: A vesiculobullous eruption caused by Coxsackie virus A6. Lancet Infect. Dis. 2014, 14, 83–86. [Google Scholar] [CrossRef]
- Lott, J.P.; Liu, K.; Landry, M.L.; Nix, W.A.; Oberste, M.S.; Bolognia, J.; King, B. Atypical hand-foot-and-mouth disease associated with coxsackievirus A6 infection. J. Am. Acad. Dermatol. 2013, 69, 736–741. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Huang, L.M.; Lu, C.Y.; Cheng, A.L.; Chang, L.Y. Atypical hand-foot-mouth disease in children: A hospital-based prospective cohort study. Virol. J. 2013, 10, 209. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CV-A6/TH Strains | Age (years) | Sex | Sample | Collection Date | Subclade | Nearest CV-A6 Strain in Structural Region (Accession No.) | Similarity with the Nearest CV-A6 Strain (% nt) | GenBank Accession No. |
|---|---|---|---|---|---|---|---|---|
| CU3482 | 1 | M | Stool | 9-Sep-19 | D3/A | China/2018 (MN845848) | 99.5 | OP896719 |
| CU3494 | 6 | F | Throat swab | 18-Sep-19 | D3/Y | China/2017 (MN845834) | 98.5 | OP896715 |
| CU3495 | 3 | F | Throat swab | 21-Sep-19 | D3/A | China/2018 (OL839942) | 99.2 | OP896717 |
| CU3496 | 3 | M | Throat swab | 23-Sep-19 | D3/Y | China/2017 (MN845834) | 98.4 | OP896716 |
| CU3510 | 8 | M | Throat swab | 8-Oct-19 | D3/A | China/2018 (OL839942) | 99.2 | OP896718 |
| CU3514 | 1 | M | Throat swab | 18-Oct-19 | D3/Y | China/2017 (MN845834) | 98.6 | OP896714 |
| CU3547 | 3 | M | Stool | 24-Jan-20 | D3/Y | China/2017 (MN845834) | 98.3 | OP896713 |
| CU3774 | 1 | M | Stool | 22-Jun-22 | D3/A | China/2018 (OL839942) | 98.0 | OP896722 |
| CU3780 | 3 | M | Throat swab | 1-Jul-22 | D3/A | China/2018 (OL839942) | 98.0 | OP896725 |
| CU3786 | 4 | F | Throat swab | 1-Jul-22 | D3/A | China/2018 (OL839942) | 98.0 | OP896729 |
| CU3792 | 1 | M | Nasal swab | 5-Jul-22 | D3/A | China/2018 (OL839942) | 98.0 | OP896728 |
| CU3812 | 2 | M | Throat swab | 1-Aug-22 | D3/A | China/2018 (OL839942) | 98.0 | OP896726 |
| CU3835 | 3 | M | Throat swab | 31-Aug-22 | D3/A | China/2018 (OL839942) | 97.9 | OP896727 |
| CU3846 | 2 | M | Throat swab | 8-Sep-22 | D3/A | China/2018 (OL839942) | 97.9 | OP896724 |
| CU3847 | 3 | F | Throat swab | 8-Sep-22 | D3/A | China/2018 (OL839942) | 97.7 | OP896723 |
| CU3913 | 3 | F | Throat swab | 20-Sep-22 | D3/A | China/2018 (OL839942) | 97.9 | OP896730 |
| CU3919 | 6 | M | Throat swab | 20-Sep-22 | D3/A | China/2018 (OL839942) | 97.7 | OP896731 |
| CU3924 | 4 | M | Throat swab | 21-Sep-22 | D3/A | China/2018 (OL839942) | 97.7 | OP896721 |
| CU3925 | 1 | M | Throat swab | 21-Sep-22 | D3/A | China/2018 (MN845848) | 98.5 | OP896720 |
| CU3933 | 4 | M | Throat swab | 21-Sep-22 | D3/A | China/2018 (OL839942) | 97.7 | OP896732 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Puenpa, J.; Saengdao, N.; Khanarat, N.; Korkong, S.; Chansaenroj, J.; Yorsaeng, R.; Wanlapakorn, N.; Poovorawan, Y. Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022. Viruses 2023, 15, 73. https://doi.org/10.3390/v15010073
Puenpa J, Saengdao N, Khanarat N, Korkong S, Chansaenroj J, Yorsaeng R, Wanlapakorn N, Poovorawan Y. Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022. Viruses. 2023; 15(1):73. https://doi.org/10.3390/v15010073
Chicago/Turabian StylePuenpa, Jiratchaya, Nutsada Saengdao, Nongkanok Khanarat, Sumeth Korkong, Jira Chansaenroj, Ritthideach Yorsaeng, Nasamon Wanlapakorn, and Yong Poovorawan. 2023. "Evolutionary and Genetic Recombination Analyses of Coxsackievirus A6 Variants Associated with Hand, Foot, and Mouth Disease Outbreaks in Thailand between 2019 and 2022" Viruses 15, no. 1: 73. https://doi.org/10.3390/v15010073