
Heparanase-1: From Cancer Biology to a Future Antiviral Target



Abstract
:1. Introduction
2. Implication of Heparanase-1 in Carcinogenesis
2.1. Primary Tumor Formation
2.2. Tumor Microenvironment Modulation
2.3. Neo-Angiogenesis Induction
2.4. Immune Response Modulation and Fibrosis Enhancement
2.5. Metastasis
3. Heparanase-1 Inhibitors for Anti-Cancer Therapy
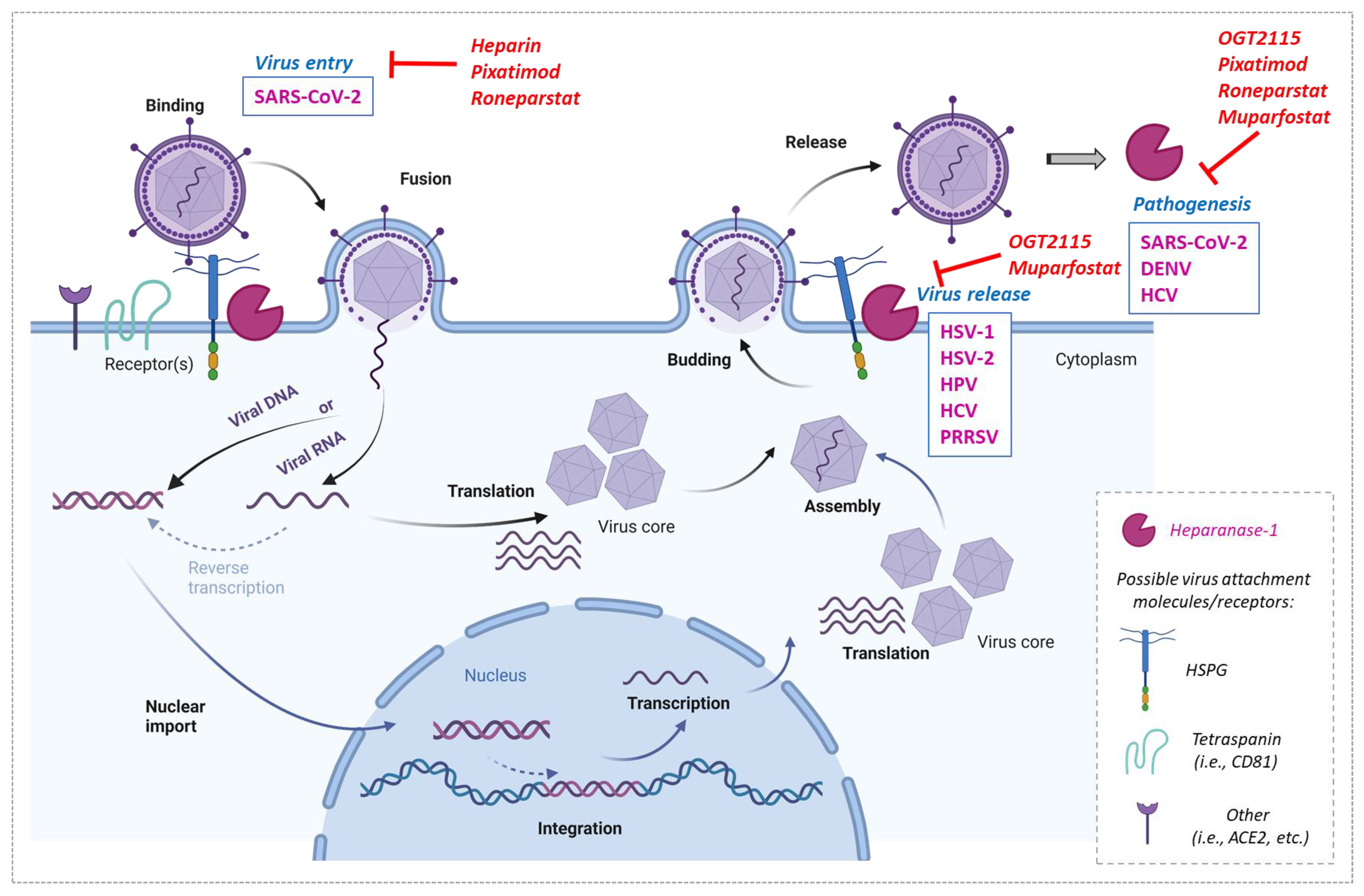
4. Heparanase-1 Involvement In Virus Infections and Their Inhibition
4.1. Herpes Simplex Virus 1 and 2 (HSV-1, HSV-2)
4.2. Human Papillomavirus (HPV)
4.3. Dengue Virus (DENV)
4.4. Hepatitis C Virus (HCV)
4.5. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2)
4.6. Porcine Reproductive and Respiratory Syndrome Virus (PPRSV)
5. Heparanase-1 Inhibitors as Future Antivirals?
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vlodavsky, I.; Singh, P.; Boyango, I.; Gutter-Kapon, L.; Elkin, M.; Sanderson, R.D.; Ilan, N. Heparanase: From Basic Research to Therapeutic Applications in Cancer and Inflammation. Drug Resist. Updates 2016, 29, 54–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlodavsky, I.; Beckhove, P.; Lerner, I.; Pisano, C.; Meirovitz, A.; Ilan, N.; Elkin, M. Significance of Heparanase in Cancer and Inflammation. Cancer Microenviron. 2012, 5, 115–132. [Google Scholar] [CrossRef] [Green Version]
- Goldshmidt, O.; Zcharia, E.; Cohen, M.; Aingorn, H.; Cohen, I.; Nadav, L.; Katz, B.-Z.; Geiger, B.; Vlodavsky, I. Heparanase Mediates Cell Adhesion Independent of Its Enzymatic Activity. FASEB J. 2003, 17, 1015–1025. [Google Scholar] [CrossRef] [Green Version]
- Ramani, V.C.; Purushothaman, A.; Stewart, M.D.; Thompson, C.A.; Vlodavsky, I.; Au, J.L.-S.; Sanderson, R.D. The Heparanase/Syndecan-1 Axis in Cancer: Mechanisms and Therapies. FEBS J. 2013, 280, 2294–2306. [Google Scholar] [CrossRef] [Green Version]
- Bashkin, P.; Doctrow, S.; Klagsbrun, M.; Svahn, C.M.; Folkman, J.; Vlodavsky, I. Basic Fibroblast Growth Factor Binds to Subendothelial Extracellular Matrix and Is Released by Heparitinase and Heparin-like Molecules. Biochemistry 1989, 28, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Vlodavsky, I.; Eldor, A.; Haimovitz-Friedman, A.; Matzner, Y.; Ishai-Michaeli, R.; Lider, O.; Naparstek, Y.; Cohen, I.R.; Fuks, Z. Expression of Heparanase by Platelets and Circulating Cells of the Immune System: Possible Involvement in Diapedesis and Extravasation. Invasion Metastasis 1992, 12, 112–127. [Google Scholar]
- Hulett, M.D.; Freeman, C.; Hamdorf, B.J.; Baker, R.T.; Harris, M.J.; Parish, C.R. Cloning of Mammalian Heparanase, an Important Enzyme in Tumor Invasion and Metastasis. Nat. Med. 1999, 5, 803–809. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Proteomics. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Rabelink, T.J.; van den Berg, B.M.; Garsen, M.; Wang, G.; Elkin, M.; van der Vlag, J. Heparanase: Roles in Cell Survival, Extracellular Matrix Remodelling and the Development of Kidney Disease. Nat. Rev. Nephrol. 2017, 13, 201–212. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Friedmann, Y. Molecular Properties and Involvement of Heparanase in Cancer Metastasis and Angiogenesis. J. Clin. Investig. 2001, 108, 341–347. [Google Scholar] [CrossRef]
- McKenzie, E.; Tyson, K.; Stamps, A.; Smith, P.; Turner, P.; Barry, R.; Hircock, M.; Patel, S.; Barry, E.; Stubberfield, C.; et al. Cloning and Expression Profiling of Hpa2, a Novel Mammalian Heparanase Family Member. Biochem. Biophys. Res. Commun. 2000, 276, 1170–1177. [Google Scholar] [CrossRef] [PubMed]
- Baraz, L.; Haupt, Y.; Elkin, M.; Peretz, T.; Vlodavsky, I. Tumor Suppressor P53 Regulates Heparanase Gene Expression. Oncogene 2006, 25, 3939–3947. [Google Scholar] [CrossRef] [Green Version]
- de Mestre, A.M.; Khachigian, L.M.; Santiago, F.S.; Staykova, M.A.; Hulett, M.D. Regulation of Inducible Heparanase Gene Transcription in Activated T Cells by Early Growth Response 1. J. Biol. Chem. 2003, 278, 50377–50385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Mestre, A.M.; Staykova, M.A.; Hornby, J.R.; Willenborg, D.O.; Hulett, M.D. Expression of the Heparan Sulfate-Degrading Enzyme Heparanase Is Induced in Infiltrating CD4+ T Cells in Experimental Autoimmune Encephalomyelitis and Regulated at the Level of Transcription by Early Growth Response Gene 1. J. Leukoc. Biol. 2007, 82, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Ostrovsky, O.; Shimoni, A.; Baryakh, P.; Morgulis, Y.; Mayorov, M.; Beider, K.; Shteingauz, A.; Ilan, N.; Vlodavsky, I.; Nagler, A. Modification of Heparanase Gene Expression in Response to Conditioning and LPS Treatment: Strong Correlation to Rs4693608 SNP. J. Leukoc. Biol. 2014, 95, 677–688. [Google Scholar] [CrossRef] [Green Version]
- Goshen, R.; Hochberg, A.A.; Korner, G.; Levy, E.; Ishai-Michaeli, R.; Elkin, M.; de Groot, N.; Vlodavsky, I. Purification and Characterization of Placental Heparanase and Its Expression by Cultured Cytotrophoblasts. Mol. Hum. Reprod. 1996, 2, 679–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, M.; Irimura, T.; Nicolson, G.L. Heparanases and Tumor Metastasis. J. Cell. Biochem. 1988, 36, 157–167. [Google Scholar] [CrossRef]
- Vlodavsky, I.; Mohsen, M.; Lider, O.; Svahn, C.M.; Ekre, H.P.; Vigoda, M.; Ishai-Michaeli, R.; Peretz, T. Inhibition of Tumor Metastasis by Heparanase Inhibiting Species of Heparin. Invasion Metastasis 1994, 14, 290–302. [Google Scholar]
- Vlodavsky, I.; Friedmann, Y.; Elkin, M.; Aingorn, H.; Atzmon, R.; Ishai-Michaeli, R.; Bitan, M.; Pappo, O.; Peretz, T.; Michal, I.; et al. Mammalian Heparanase: Gene Cloning, Expression and Function in Tumor Progression and Metastasis. Nat. Med. 1999, 5, 793–802. [Google Scholar] [CrossRef]
- Parish, C.R.; Freeman, C.; Hulett, M.D. Heparanase: A Key Enzyme Involved in Cell Invasion. Biochim. Biophys. Acta 2001, 1471, M99–M108. [Google Scholar] [CrossRef]
- Gohji, K.; Okamoto, M.; Kitazawa, S.; Toyoshima, M.; Dong, J.; Katsuoka, Y.; Nakajima, M. Heparanase Protein and Gene Expression in Bladder Cancer. J. Urol. 2001, 166, 1286–1290. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Xu, S.; Tan, Q.; Liu, L. High Expression of Heparanase-2 Is an Independent Prognostic Parameter for Favorable Survival in Gastric Cancer Patients. Cancer Epidemiol. 2013, 37, 1010–1013. [Google Scholar] [CrossRef]
- Sato, T.; Yamaguchi, A.; Goi, T.; Hirono, Y.; Takeuchi, K.; Katayama, K.; Matsukawa, S. Heparanase Expression in Human Colorectal Cancer and Its Relationship to Tumor Angiogenesis, Hematogenous Metastasis, and Prognosis. J. Surg. Oncol. 2004, 87, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Chen, X.-P.; Wu, M.-S.; Cui, W.; Zhong, M. Expressions of Heparanase and Upstream Stimulatory Factor in Hepatocellular Carcinoma. Eur. J. Med. Res. 2014, 19, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinyo, Y.; Kodama, J.; Hongo, A.; Yoshinouchi, M.; Hiramatsu, Y. Heparanase Expression Is an Independent Prognostic Factor in Patients with Invasive Cervical Cancer. Ann. Oncol. 2003, 14, 1505–1510. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-J.; Shi, L.; Wang, X.-M.; Yang, G.-W. Heparanase Is a Novel Biomarker for Immune Infiltration and Prognosis in Breast Cancer. Aging 2021, 13, 20836–20852. [Google Scholar] [CrossRef]
- Bitan, M.; Polliack, A.; Zecchina, G.; Nagler, A.; Friedmann, Y.; Nadav, L.; Deutsch, V.; Pecker, I.; Eldor, A.; Vlodavsky, I.; et al. Heparanase Expression in Human Leukemias Is Restricted to Acute Myeloid Leukemias. Exp. Hematol. 2002, 30, 34–41. [Google Scholar] [CrossRef]
- Ramani, V.C.; Zhan, F.; He, J.; Barbieri, P.; Noseda, A.; Tricot, G.; Sanderson, R.D. Targeting Heparanase Overcomes Chemoresistance and Diminishes Relapse in Myeloma. Oncotarget 2016, 7, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Galli, M.; Chatterjee, M.; Grasso, M.; Specchia, G.; Magen, H.; Einsele, H.; Celeghini, I.; Barbieri, P.; Paoletti, D.; Pace, S.; et al. Phase I Study of the Heparanase Inhibitor Roneparstat: An Innovative Approach for Ultiple Myeloma Therapy. Haematologica 2018, 103, e469–e472. [Google Scholar] [CrossRef]
- Purushothaman, A.; Sanderson, R.D. Heparanase: A Dynamic Promoter of Myeloma Progression. Adv. Exp. Med. Biol. 2020, 1221, 331–349. [Google Scholar] [CrossRef]
- Rangarajan, S.; Tripathi, K.; Bandari, S.K.; Brown, E.E.; Sanderson, R.D. Heparanase in Myeloma Related Renal Dysfunction: Role in Promoting Nephrotoxicity and Potential as a Novel Biomarker for Early Detection. J. Clin. Oncol. 2020, 38, e20554. [Google Scholar] [CrossRef]
- Putz, E.M.; Mayfosh, A.J.; Kos, K.; Barkauskas, D.S.; Nakamura, K.; Town, L.; Goodall, K.J.; Yee, D.Y.; Poon, I.K.; Baschuk, N.; et al. NK Cell Heparanase Controls Tumor Invasion and Immune Surveillance. J. Clin. Investig. 2017, 127, 2777–2788. [Google Scholar] [CrossRef]
- Dang, C.V. MYC on the Path to Cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontomanolis, E.N.; Koutras, A.; Syllaios, A.; Schizas, D.; Mastoraki, A.; Garmpis, N.; Diakosavvas, M.; Angelou, K.; Tsatsaris, G.; Pagkalos, A.; et al. Role of Oncogenes and Tumor-Suppressor Genes in Carcinogenesis: A Review. Anticancer Res. 2020, 40, 6009–6015. [Google Scholar] [CrossRef] [PubMed]
- Boyango, I.; Barash, U.; Naroditsky, I.; Li, J.-P.; Hammond, E.; Ilan, N.; Vlodavsky, I. Heparanase Cooperates with Ras to Drive Breast and Skin Tumorigenesis. Cancer Res. 2014, 74, 4504–4514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, G.; Liu, D.; Xing, M.; Tauler, J.; Prinz, R.A.; Xu, X. Induction of Heparanase-1 Expression by Mutant B-Raf Kinase: Role of GA Binding Protein in Heparanase-1 Promoter Activation. Neoplasia 2010, 12, 946–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Xie, R.; Qin, Y.; Xiao, Y.-F.; Yong, X.; Zheng, L.; Dong, H.; Yang, S.-M. Human Telomerase Reverse Transcriptase (HTERT) Promotes Gastric Cancer Invasion through Cooperating with c-Myc to Upregulate Heparanase Expression. Oncotarget 2015, 7, 11364–11379. [Google Scholar] [CrossRef]
- Amin, R.; Tripathi, K.; Sanderson, R.D. Nuclear Heparanase Regulates Chromatin Remodeling, Gene Expression and PTEN Tumor Suppressor Function. Cells 2020, 9, 2038. [Google Scholar] [CrossRef]
- Turner, N.; Grose, R. Fibroblast Growth Factor Signalling: From Development to Cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Reiland, J.; Kempf, D.; Roy, M.; Denkins, Y.; Marchetti, D. FGF2 Binding, Signaling, and Angiogenesis Are Modulated by Heparanase in Metastatic Melanoma Cells. Neoplasia 2006, 8, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Escobar Galvis, M.L.; Jia, J.; Zhang, X.; Jastrebova, N.; Spillmann, D.; Gottfridsson, E.; van Kuppevelt, T.H.; Zcharia, E.; Vlodavsky, I.; Lindahl, U.; et al. Transgenic or Tumor-Induced Expression of Heparanase Upregulates Sulfation of Heparan Sulfate. Nat. Chem. Biol. 2007, 3, 773–778. [Google Scholar] [CrossRef] [PubMed]
- Marques, C.; Reis, C.A.; Vivès, R.R.; Magalhães, A. Heparan Sulfate Biosynthesis and Sulfation Profiles as Modulators of Cancer Signalling and Progression. Front. Oncol. 2021, 11, 778752. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Gambaro, G.; Tibaldi, E.; Brunati, A.M.; Gastaldello, A.; D’Angelo, A.; Onisto, M.; Lupo, A. Heparanase and Syndecan-1 Interplay Orchestrates Fibroblast Growth Factor-2-Induced Epithelial-Mesenchymal Transition in Renal Tubular Cells. J. Biol. Chem. 2012, 287, 1478–1488. [Google Scholar] [CrossRef] [Green Version]
- Yehya, A.H.S.; Asif, M.; Petersen, S.H.; Subramaniam, A.V.; Kono, K.; Majid, A.M.S.A.; Oon, C.E. Angiogenesis: Managing the Culprits behind Tumorigenesis and Metastasis. Medicina 2018, 54, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saman, H.; Raza, S.S.; Uddin, S.; Rasul, K. Inducing Angiogenesis, a Key Step in Cancer Vascularization, and Treatment Approaches. Cancers 2020, 12, 1172. [Google Scholar] [CrossRef]
- Zetser, A.; Bashenko, Y.; Edovitsky, E.; Levy-Adam, F.; Vlodavsky, I.; Ilan, N. Heparanase Induces Vascular Endothelial Growth Factor Expression: Correlation with P38 Phosphorylation Levels and Src Activation. Cancer Res. 2006, 66, 1455–1463. [Google Scholar] [CrossRef] [Green Version]
- Cohen-Kaplan, V.; Naroditsky, I.; Zetser, A.; Ilan, N.; Vlodavsky, I.; Doweck, I. Heparanase Induces VEGF C and Facilitates Tumor Lymphangiogenesis. Int. J. Cancer 2008, 123, 2566–2573. [Google Scholar] [CrossRef] [Green Version]
- Hermano, E.; Meirovitz, A.; Meir, K.; Nussbaum, G.; Appelbaum, L.; Peretz, T.; Elkin, M. Macrophage Polarization in Pancreatic Carcinoma: Role of Heparanase Enzyme. J. Natl. Cancer Inst. 2014, 106, dju332. [Google Scholar] [CrossRef] [Green Version]
- Thompson, C.A.; Purushothaman, A.; Ramani, V.C.; Vlodavsky, I.; Sanderson, R.D. Heparanase Regulates Secretion, Composition, and Function of Tumor Cell-Derived Exosomes. J. Biol. Chem. 2013, 288, 10093–10099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shteingauz, A.; Boyango, I.; Naroditsky, I.; Hammond, E.; Gruber, M.; Doweck, I.; Ilan, N.; Vlodavsky, I. Heparanase Enhances Tumor Growth and Chemoresistance by Promoting Autophagy. Cancer Res. 2015, 75, 3946–3957. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting Tumor-Associated Macrophages to Synergize Tumor Immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Masola, V.; Zaza, G.; Secchi, M.F.; Gambaro, G.; Lupo, A.; Onisto, M. Heparanase Is a Key Player in Renal Fibrosis by Regulating TGF-β Expression and Activity. Biochim. Biophys. Acta 2014, 1843, 2122–2128. [Google Scholar] [CrossRef] [PubMed]
- Caruana, I.; Savoldo, B.; Hoyos, V.; Weber, G.; Liu, H.; Kim, E.S.; Ittmann, M.M.; Marchetti, D.; Dotti, G. Heparanase Promotes Tumor Infiltration and Antitumor Activity of CAR-Redirected T Lymphocytes. Nat. Med. 2015, 21, 524–529. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, Y.; Miyake, M.; Shimada, K.; Fujii, T.; Hori, S.; Morizawa, Y.; Nakai, Y.; Anai, S.; Tanaka, N.; Konishi, N.; et al. Inhibition of Heparanase Expression Results in Suppression of Invasion, Migration and Adhesion Abilities of Bladder Cancer Cells. Int. J. Mol. Sci. 2020, 21, 3789. [Google Scholar] [CrossRef] [PubMed]
- Edovitsky, E.; Elkin, M.; Zcharia, E.; Peretz, T.; Vlodavsky, I. Heparanase Gene Silencing, Tumor Invasiveness, Angiogenesis, and Metastasis. J. Natl. Cancer Inst. 2004, 96, 1219–1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zcharia, E.; Jia, J.; Zhang, X.; Baraz, L.; Lindahl, U.; Peretz, T.; Vlodavsky, I.; Li, J.-P. Newly Generated Heparanase Knock-out Mice Unravel Co-Regulation of Heparanase and Matrix Metalloproteinases. PLoS ONE 2009, 4, e5181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Viola, C.M.; Brzozowski, A.M.; Davies, G.J. Structural Characterization of Human Heparanase Reveals Insights into Substrate Recognition. Nat. Struct. Mol. Biol. 2015, 22, 1016–1022. [Google Scholar] [CrossRef] [Green Version]
- de Boer, C.; Armstrong, Z.; Lit, V.A.J.; Barash, U.; Ruijgrok, G.; Boyango, I.; Weitzenberg, M.M.; Schröder, S.P.; Sarris, A.J.C.; Meeuwenoord, N.J.; et al. Mechanism-Based Heparanase Inhibitors Reduce Cancer Metastasis in vivo. Proc. Natl. Acad. Sci. USA 2022, 119, e2203167119. [Google Scholar] [CrossRef]
- Casu, B.; Vlodavsky, I.; Sanderson, R.D. Non-Anticoagulant Heparins and Inhibition of Cancer. Pathophysiol. Haemost. Thromb. 2008, 36, 195–203. [Google Scholar] [CrossRef]
- Jia, L.; Ma, S. Recent Advances in the Discovery of Heparanase Inhibitors as Anti-Cancer Agents. Eur. J. Med. Chem. 2016, 121, 209–220. [Google Scholar] [CrossRef]
- Noseda, A.; Barbieri, P. Roneparstat: Development, Preclinical and Clinical Studies. Adv. Exp. Med. Biol. 2020, 1221, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Guimond, S.E.; Mycroft-West, C.J.; Gandhi, N.S.; Tree, J.A.; Le, T.T.; Spalluto, C.M.; Humbert, M.V.; Buttigieg, K.R.; Coombes, N.; Elmore, M.J.; et al. Synthetic Heparan Sulfate Mimetic Pixatimod (PG545) Potently Inhibits SARS-CoV-2 By Disrupting The Spike-ACE2 Interaction. ACS Cent. Sci. 2022, 8, 527–545. [Google Scholar] [CrossRef]
- Dredge, K.; Brennan, T.V.; Hammond, E.; Lickliter, J.D.; Lin, L.; Bampton, D.; Handley, P.; Lankesheer, F.; Morrish, G.; Yang, Y.; et al. A Phase I Study of the Novel Immunomodulatory Agent PG545 (Pixatimod) in Subjects with Advanced Solid Tumours. Br. J. Cancer 2018, 118, 1035–1041. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.-J.; Lee, P.-H.; Han, K.-H.; Fan, J.; Cheung, T.T.; Hu, R.-H.; Paik, S.W.; Lee, W.-C.; Chau, G.-Y.; Jeng, L.-B.; et al. A Phase III Trial of Muparfostat (PI-88) as Adjuvant Therapy in Patients with Hepatitis Virus Related Hepatocellular Carcinoma (HV-HCC) after Resection. Ann. Oncol. 2017, 28, v213. [Google Scholar] [CrossRef]
- Khasraw, M.; Pavlakis, N.; McCowatt, S.; Underhill, C.; Begbie, S.; de Souza, P.; Boyce, A.; Parnis, F.; Lim, V.; Harvie, R.; et al. Multicentre Phase I/II Study of PI-88, a Heparanase Inhibitor in Combination with Docetaxel in Patients with Metastatic Castrate-Resistant Prostate Cancer. Ann. Oncol. 2010, 21, 1302–1307. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.D.; Robinson, W.A.; Millward, M.J.; Powell, A.; Price, T.J.; Thomson, D.B.; Walpole, E.T.; Haydon, A.M.; Creese, B.R.; Roberts, K.L.; et al. A Phase II Study of the Heparanase Inhibitor PI-88 in Patients with Advanced Melanoma. Investig. New Drugs 2008, 26, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-J.; Lee, P.-H.; Lin, D.-Y.; Wu, C.-C.; Jeng, L.-B.; Lin, P.-W.; Mok, K.-T.; Lee, W.-C.; Yeh, H.-Z.; Ho, M.-C.; et al. Heparanase Inhibitor PI-88 as Adjuvant Therapy for Hepatocellular Carcinoma after Curative Resection: A Randomized Phase II Trial for Safety and Optimal Dosage. J. Hepatol. 2009, 50, 958–968. [Google Scholar] [CrossRef]
- Karoli, T.; Liu, L.; Fairweather, J.K.; Hammond, E.; Li, C.P.; Cochran, S.; Bergefall, K.; Trybala, E.; Addison, R.S.; Ferro, V. Synthesis, Biological Activity, and Preliminary Pharmacokinetic Evaluation of Analogues of a Phosphosulfomannan Angiogenesis Inhibitor (PI-88). J. Med. Chem. 2005, 48, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- Courtney, S.M.; Hay, P.A.; Buck, R.T.; Colville, C.S.; Phillips, D.J.; Scopes, D.I.C.; Pollard, F.C.; Page, M.J.; Bennett, J.M.; Hircock, M.L.; et al. Furanyl-1,3-Thiazol-2-Yl and Benzoxazol-5-Yl Acetic Acid Derivatives: Novel Classes of Heparanase Inhibitor. Bioorg. Med. Chem. Lett. 2005, 15, 2295–2299. [Google Scholar] [CrossRef]
- Pan, W.; Miao, H.-Q.; Xu, Y.-J.; Navarro, E.C.; Tonra, J.R.; Corcoran, E.; Lahiji, A.; Kussie, P.; Kiselyov, A.S.; Wong, W.C.; et al. 1-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-3-[4-(1H-Benzoimidazol-2-Yl)-Phenyl]-Urea Derivatives as Small Molecule Heparanase Inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 409–412. [Google Scholar] [CrossRef]
- Weissmann, M.; Arvatz, G.; Horowitz, N.; Feld, S.; Naroditsky, I.; Zhang, Y.; Ng, M.; Hammond, E.; Nevo, E.; Vlodavsky, I.; et al. Heparanase-Neutralizing Antibodies Attenuate Lymphoma Tumor Growth and Metastasis. Proc. Natl. Acad. Sci. USA 2016, 113, 704–709. [Google Scholar] [CrossRef] [Green Version]
- Mohan, C.D.; Hari, S.; Preetham, H.D.; Rangappa, S.; Barash, U.; Ilan, N.; Nayak, S.C.; Gupta, V.K.; Vlodavsky, I.; Rangappa, K.S. Targeting Heparanase in Cancer: Inhibition by Synthetic, Chemically Modified, and Natural Compounds. iScience 2019, 15, 360–390. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Hadigal, S.R.; Jaishankar, D.; Shukla, D. Viral Activation of Heparanase Drives Pathogenesis of Herpes Simplex Virus-1. Cell Rep. 2017, 20, 439–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadigal, S.R.; Agelidis, A.M.; Karasneh, G.A.; Antoine, T.E.; Yakoub, A.M.; Ramani, V.C.; Djalilian, A.R.; Sanderson, R.D.; Shukla, D. Heparanase Is a Host Enzyme Required for Herpes Simplex Virus-1 Release from Cells. Nat. Commun. 2015, 6, 6985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hopkins, J.; Yadavalli, T.; Agelidis, A.M.; Shukla, D. Host Enzymes Heparanase and Cathepsin L Promote Herpes Simplex Virus 2 Release from Cells. J. Virol. 2018, 92, e01179-18. [Google Scholar] [CrossRef] [Green Version]
- Puerta-Guardo, H.; Glasner, D.R.; Harris, E. Dengue Virus NS1 Disrupts the Endothelial Glycocalyx, Leading to Hyperpermeability. PLoS Pathog. 2016, 12, e1005738. [Google Scholar] [CrossRef] [Green Version]
- Surviladze, Z.; Sterkand, R.T.; Ozbun, M.A. Interaction of Human Papillomavirus Type 16 Particles with Heparan Sulfate and Syndecan-1 Molecules in the Keratinocyte Extracellular Matrix Plays an Active Role in Infection. J. Gen. Virol. 2015, 96, 2232–2241. [Google Scholar] [CrossRef]
- Guo, C.; Zhu, Z.; Guo, Y.; Wang, X.; Yu, P.; Xiao, S.; Chen, Y.; Cao, Y.; Liu, X. Heparanase Upregulation Contributes to Porcine Reproductive and Respiratory Syndrome Virus Release. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Gallard, C.; Lebsir, N.; Khursheed, H.; Reungoat, E.; Plissonnier, M.-L.; Bré, J.; Michelet, M.; Chouik, Y.; Zoulim, F.; Pécheur, E.-I.; et al. Heparanase-1 Is Upregulated by Hepatitis C Virus and Favors Its Replication. J. Hepatol. 2022, 77, 29–41. [Google Scholar] [CrossRef]
- Mycroft-West, C.J.; Su, D.; Pagani, I.; Rudd, T.R.; Elli, S.; Gandhi, N.S.; Guimond, S.E.; Miller, G.J.; Meneghetti, M.C.Z.; Nader, H.B.; et al. Heparin Inhibits Cellular Invasion by SARS-CoV-2: Structural Dependence of the Interaction of the Spike S1 Receptor-Binding Domain with Heparin. Thromb. Haemost. 2020, 120, 1700–1715. [Google Scholar] [CrossRef]
- Xiang, J.; Lu, M.; Shi, M.; Cheng, X.; Kwakwa, K.A.; Davis, J.L.; Su, X.; Bakewell, S.J.; Zhang, Y.; Fontana, F.; et al. Heparanase Blockade as a Novel Dual-Targeting Therapy for COVID-19. J. Virol. 2022, 96, e0005722. [Google Scholar] [CrossRef]
- WHO. Herpes Simplex Virus. 2022. Available online: https://www.who.int/news-room/fact-sheets/detail/herpes-simplex-virus (accessed on 2 December 2022).
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes Simplex Virus: Global Infection Prevalence and Incidence Estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Steiner, I.; Kennedy, P.G.E.; Pachner, A.R. The Neurotropic Herpes Viruses: Herpes Simplex and Varicella-Zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Whitley, R.; Baines, J. Clinical Management of Herpes Simplex Virus Infections: Past, Present, and Future. F1000Research 2018, 7, F1000 Faculty Rev-1726. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, C.D.; Shukla, D. The Importance of Heparan Sulfate in Herpesvirus Infection. Virol. Sin. 2008, 23, 383–393. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kapoor, D.; Shukla, D. Role of Heparanase and Syndecan-1 in HSV-1 Release from Infected Cells. Viruses 2022, 14, 2156. [Google Scholar] [CrossRef]
- Said, J.S.; Trybala, E.; Görander, S.; Ekblad, M.; Liljeqvist, J.-Å.; Jennische, E.; Lange, S.; Bergström, T. The Cholestanol-Conjugated Sulfated Oligosaccharide PG545 Disrupts the Lipid Envelope of Herpes Simplex Virus Particles. Antimicrob. Agents Chemother. 2016, 60, 1049–1057. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Viruses | Role of Heparanase-1 in Infection/Pathogenesis | Heparanase Inhibitors Blocking Infection | References |
|---|---|---|---|
| Herpes simplex virus (HSV-1) | Increased virus release. ECM damage leading to disease pathologies | OGT2115 | [73,74] |
| Herpes simplex virus (HSV-2) | Increased virus release | OGT2115 Pixatimod (PG545) | [75] |
| Dengue virus (DENV) | Increase in severity of disease symptoms | OGT2115 | [76] |
| Human papilloma virus (HPV) | Increased virus release | OGT2115 | [77] |
| Porcine respiratory and reproductive syncytial virus (PRRSV) | Increased virus release | N/A | [78] |
| Hepatitis C virus (HCV) | Increased rate of infection/virus release | Muparfostat (PI-88) | [79] |
| SARS-CoV-2 | Attachment, entry | Heparin Pixatimod (PG545) Roneparstat (SST001) | [62,80,81] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lebsir, N.; Zoulim, F.; Grigorov, B. Heparanase-1: From Cancer Biology to a Future Antiviral Target. Viruses 2023, 15, 237. https://doi.org/10.3390/v15010237
Lebsir N, Zoulim F, Grigorov B. Heparanase-1: From Cancer Biology to a Future Antiviral Target. Viruses. 2023; 15(1):237. https://doi.org/10.3390/v15010237
Chicago/Turabian StyleLebsir, Nadjet, Fabien Zoulim, and Boyan Grigorov. 2023. "Heparanase-1: From Cancer Biology to a Future Antiviral Target" Viruses 15, no. 1: 237. https://doi.org/10.3390/v15010237