
Phylogenomic and Structural Analysis of the Monkeypox Virus Shows Evolution towards Increased Stability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Selection and Phylogeny
2.2. Genome Mutation Analysis
2.3. Computational Structural Analysis of the MPXV Proteins
3. Results
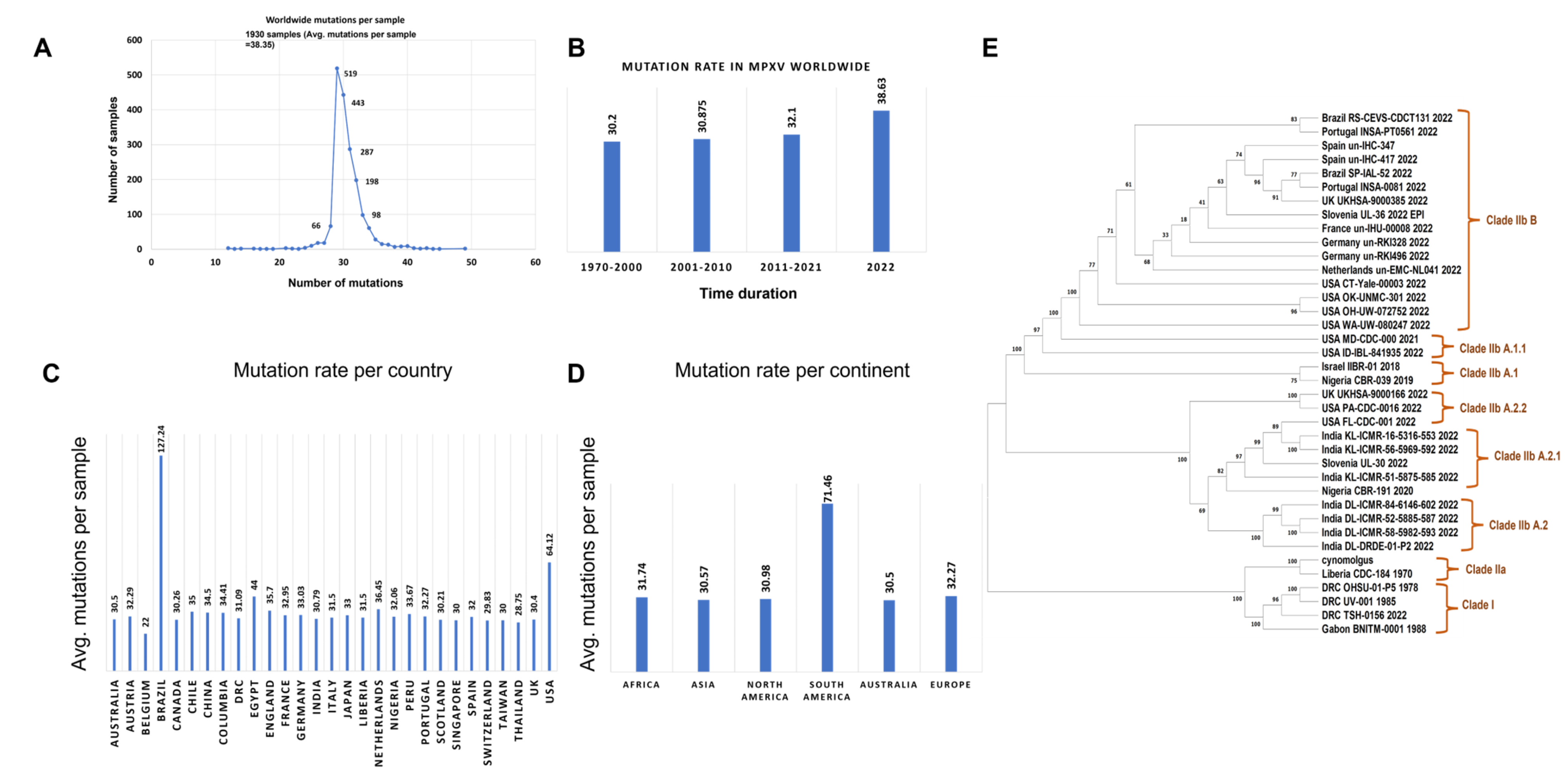
3.1. MPXV Genome Sequences from the Ongoing Outbreak Show a High Mutation Rate
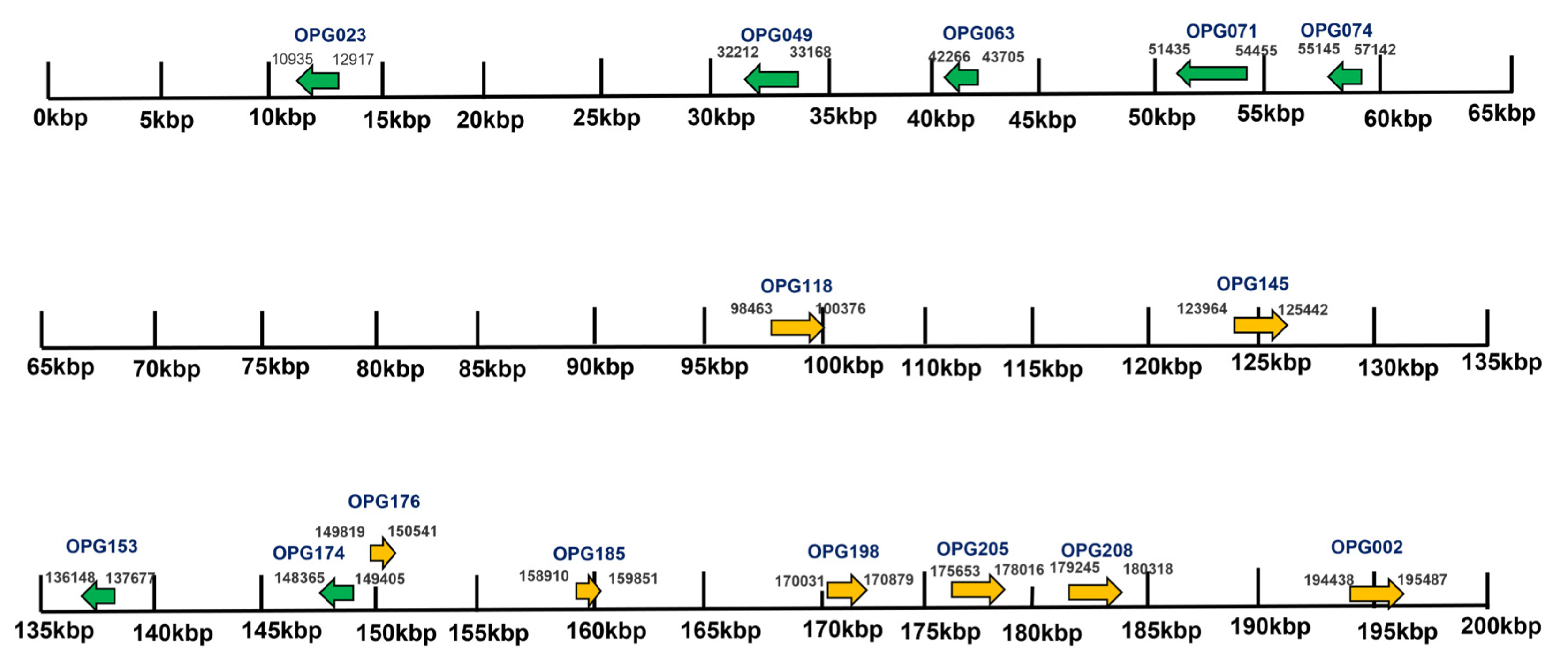
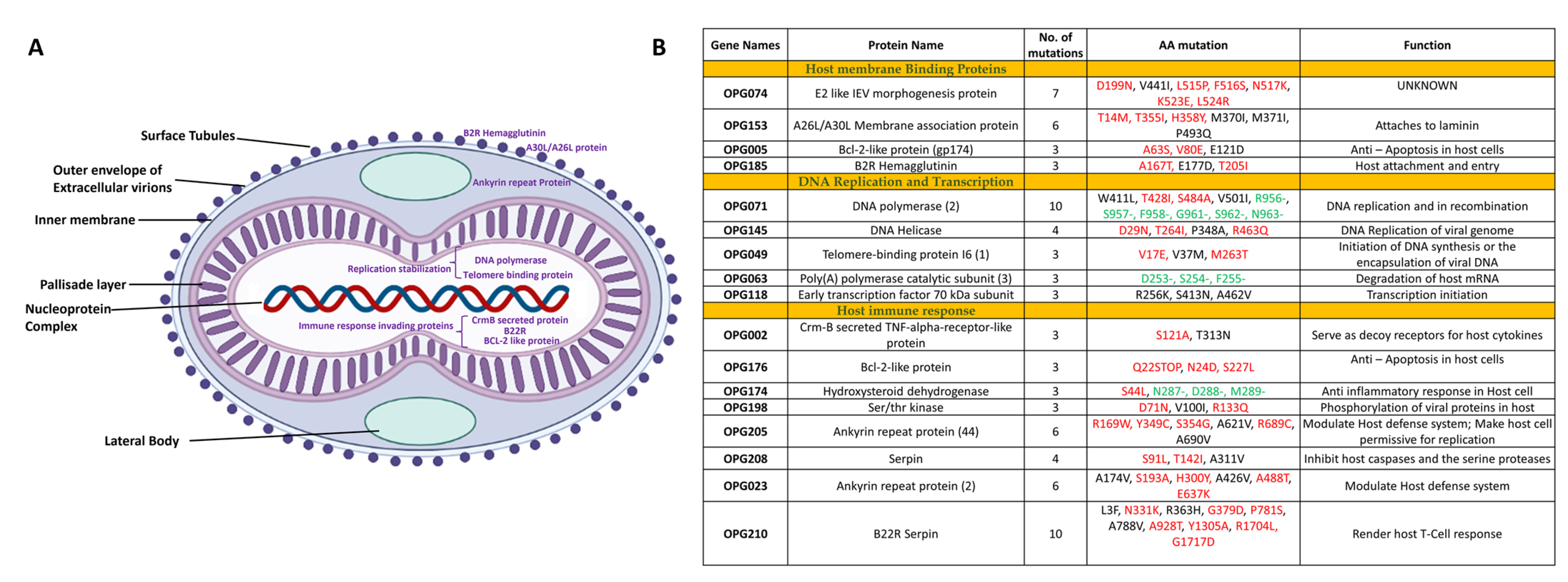
3.2. Variable End Regions of the MPXV Genome Show a Higher Number of Mutations
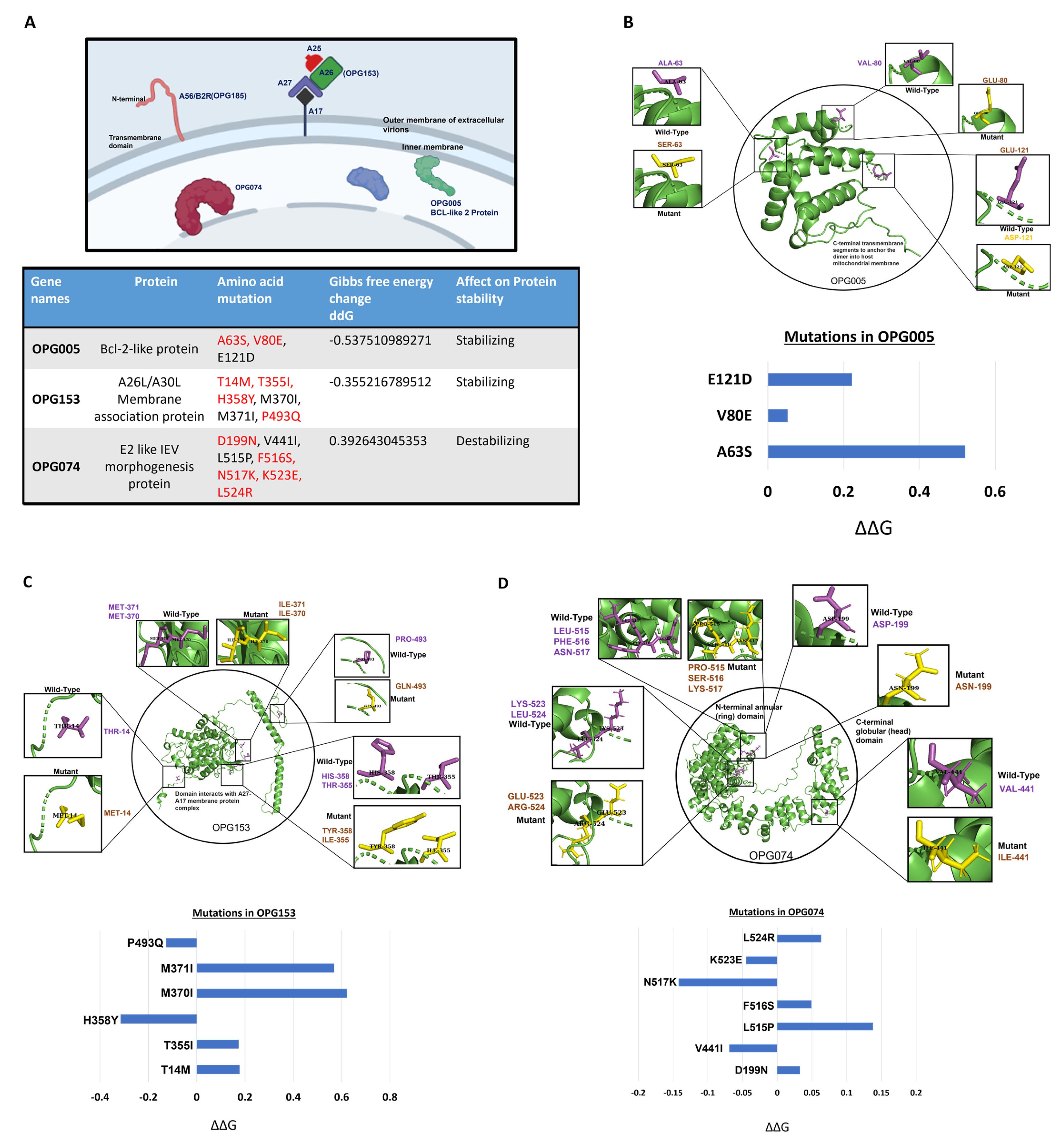
3.3. Structural Analysis of the Mutations Suggests the Greater Fitness and Infectivity of the New Variants of MPXV
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ladnyj, I.D.; Ziegler, P.; Kima, E. A human infection caused by monkeypox virus in Basankusu Territory, Democratic Republic of the Congo. Bull. World Health Organ. 1972, 46, 593–597. [Google Scholar] [PubMed]
- Breman, J.G.; Steniowski, M.V.; Zanotto, E.; Gromyko, A.I.; Arita, I. Human monkeypox, 1970–79. Bull. World Health Organ. 1980, 58, 165–182. [Google Scholar] [PubMed]
- Adler, H.; Gould, S.; Hine, P.; Snell, L.B.; Wong, W.; Houlihan, C.F.; Osborne, J.C.; Rampling, T.; Beadsworth, M.B.; Duncan, C.J.; et al. Clinical features and management of human monkeypox: A retrospective observational study in the UK. Lancet Infect. Dis. 2022, 22, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Sklenovská, N.; Van Ranst, M. Emergence of Monkeypox as the Most Important Orthopoxvirus Infection in Humans. Front. Public Health 2018, 6, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knipe, D.M.; Howley, P.M.; Griffin, D.E.; Lamb, R.A.; Martin, M.A.; Roizman, B.; Straus, S.E. Fields Virology, Moss, B., Ed.; 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 2905–2945. [Google Scholar]
- Henderson, D.A. The eradication of smallpox—An overview of the past, present, and future. Vaccine 2011, 30, 19. [Google Scholar] [CrossRef]
- McCollum, A.M.; Damon, I.K. Human monkeypox. Clin. Infect. Dis. 2014, 58, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Fine, P.E.M.; Jezek, Z.; Grab, B.; Dixon, H. The transmission potential of monkeypox virus in human populations. Int. J. Epidemiol. 1988, 17, 643–650. [Google Scholar] [CrossRef]
- Reynolds, M.G.; Damon, I.K. Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Microbiol. 2012, 20, 80–87. [Google Scholar] [CrossRef]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A tale of two clades: Monkeypox viruses. J. Gen. Virol. 2005, 86 Pt 10, 2661–2672. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. 2022 Monkeypox Outbreak Global Map. Available online: https://www.cdc.gov/poxvirus/monkeypox/response/2022/world-map.html (accessed on 20 October 2022).
- World Health Organization. Multi-Country Monkeypox Outbreak in Non-Endemic Countries. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON385 (accessed on 20 October 2022).
- Seang, S.; Burrel, S.; Todesco, E.; Leducq, V.; Monsel, G.; Le Pluart, D.; Cordevant, C.; Pourcher, V.; Palich, R. Evidence of human-to-dog transmission of monkeypox virus. Lancet J. 2022, 400, P658–P659. [Google Scholar] [CrossRef]
- Shu, Y.; McCauley, J. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, 17. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [Green Version]
- Pathan, R.K.; Biswas, M.; Khandaker, M.U. Time series prediction of COVID-19 by mutation rate analysis using recurrent neural network-based LSTM model. Chaos Solitons Fractals 2020, 138, 13. [Google Scholar] [CrossRef]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [Green Version]
- Laimer, J.; Hofer, H.; Fritz, M.; Wegenkittl, S.; Lackner, P. MAESTRO—Multi agent stability prediction upon point mutations. BMC Bioinform. 2015, 16, 116. [Google Scholar] [CrossRef] [Green Version]
- Shete, A.M.; Yadav, P.D.; Kumar, A.; Patil, S.; Patil, D.Y.; Joshi, Y.; Majumdar, T.; Relhan, V.; Sahay, R.R.; Vasu, M.; et al. Genome characterization of monkeypox cases detected in India: Identification of three sub clusters among A.2 lineage. bioRxiv 2022. bioRxiv: 2022.09.16.507742. [Google Scholar] [CrossRef]
- Hunt, R.; Sauna, Z.E.; Ambudkar, S.V.; Gottesman, M.M.; Kimchi-Sarfaty, C. Silent (Synonymous) SNPs: Should We Care About Them? In Single Nucleotide Polymorphisms: Methods and Protocols; Komar, A.A., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 23–39. [Google Scholar]
- Robert, F.; Pelletier, J. Exploring the Impact of Single-Nucleotide Polymorphisms on Translation. Front. Genet. 2018, 9, v507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, S. Why are RNA virus mutation rates so damn high? PLoS Biol. 2018, 16, e3000003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alzhanova, D.; Hammarlund, E.; Reed, J.; Meermeier, E.; Rawlings, S.; Ray, C.A.; Edwards, D.M.; Bimber, B.; Legasse, A.; Planer, S.; et al. T cell inactivation by poxviral B22 family proteins increases viral virulence. PLoS Pathog. 2014, 10, e1004123. [Google Scholar] [CrossRef] [PubMed]
- Herbert, M.H.; Squire, C.J.; A Mercer, A. Poxviral ankyrin proteins. Viruses 2015, 7, 709–738. [Google Scholar] [CrossRef] [PubMed]
- Odon, V.; Georgana, I.; Holley, J.; Morata, J.; de Motes, C.M. Novel Class of Viral Ankyrin Proteins Targeting the Host E3 Ubiquitin Ligase Cullin-2. J. Virol. 2018, 92, 01374-18. [Google Scholar] [CrossRef] [Green Version]
- Szpara, M.L.; Van Doorslaer, K. Mechanisms of DNA Virus Evolution. Encycl. Virol. 2021, 1, 71–78. [Google Scholar] [CrossRef]
- Chittenden, T. BH3 domains: Intracellular death-ligands critical for initiating apoptosis. Cancer Cell 2002, 2, 165–166. [Google Scholar] [CrossRef] [Green Version]
- Moss, B. Poxvirus Cell Entry: How Many Proteins Does it Take? Viruses 2012, 4, 688–707. [Google Scholar] [CrossRef]
- Chiu, W.-L.; Lin, C.-L.; Yang, M.-H.; Tzou, D.-L.M.; Chang, W. Vaccinia virus 4c (A26L) protein on intracellular mature virus binds to the extracellular cellular matrix laminin. J. Virol. 2007, 81, 2149–2157. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, D.; Rodriguez, J.R.; Esteban, M. The vaccinia virus 14-kilodalton fusion protein forms a stable complex with the processed protein encoded by the vaccinia virus A17L gene. J. Virol. 1993, 67, 3435–3440. [Google Scholar] [CrossRef]
- Bunge, E.M.; Hoet, B.; Chen, L.; Lienert, F.; Weidenthaler, H.; Baer, L.R.; Steffen, R. The changing epidemiology of human monkeypox—A potential threat? A systematic review. PLoS Negl. Trop. Dis. 2022, 16, e0010141. [Google Scholar] [CrossRef]
- Choudhary, O.P.; Priyanka; Chopra, H.; Shafaati, M.; Dhawan, M.; Metwally, A.A.; Saied, A.A.; Rabaan, A.A.; Alhumaid, S.; Al Mutair, A.; et al. Reverse zoonosis and its relevance to the monkeypox outbreak 2022. New Microbes New Infect. 2022, 15, 50. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yadav, P.; Devasurmutt, Y.; Tatu, U. Phylogenomic and Structural Analysis of the Monkeypox Virus Shows Evolution towards Increased Stability. Viruses 2023, 15, 127. https://doi.org/10.3390/v15010127
Yadav P, Devasurmutt Y, Tatu U. Phylogenomic and Structural Analysis of the Monkeypox Virus Shows Evolution towards Increased Stability. Viruses. 2023; 15(1):127. https://doi.org/10.3390/v15010127
Chicago/Turabian StyleYadav, Priya, Yashas Devasurmutt, and Utpal Tatu. 2023. "Phylogenomic and Structural Analysis of the Monkeypox Virus Shows Evolution towards Increased Stability" Viruses 15, no. 1: 127. https://doi.org/10.3390/v15010127