
Ebola Virus Activates IRE1α-Dependent XBP1u Splicing
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Virus Infection and Titration
2.3. Antibodies
2.4. Plasmids
2.5. UPRE Luciferase Reporter Assay
2.6. qRT-PCR Analysis
2.7. XBP1u Splicing
2.8. ATF6 Cleavage Assay
2.9. Western Blot Analysis
2.10. Indirect Immunofluorescence Analysis (IFA)
2.11. Statistical Analyses
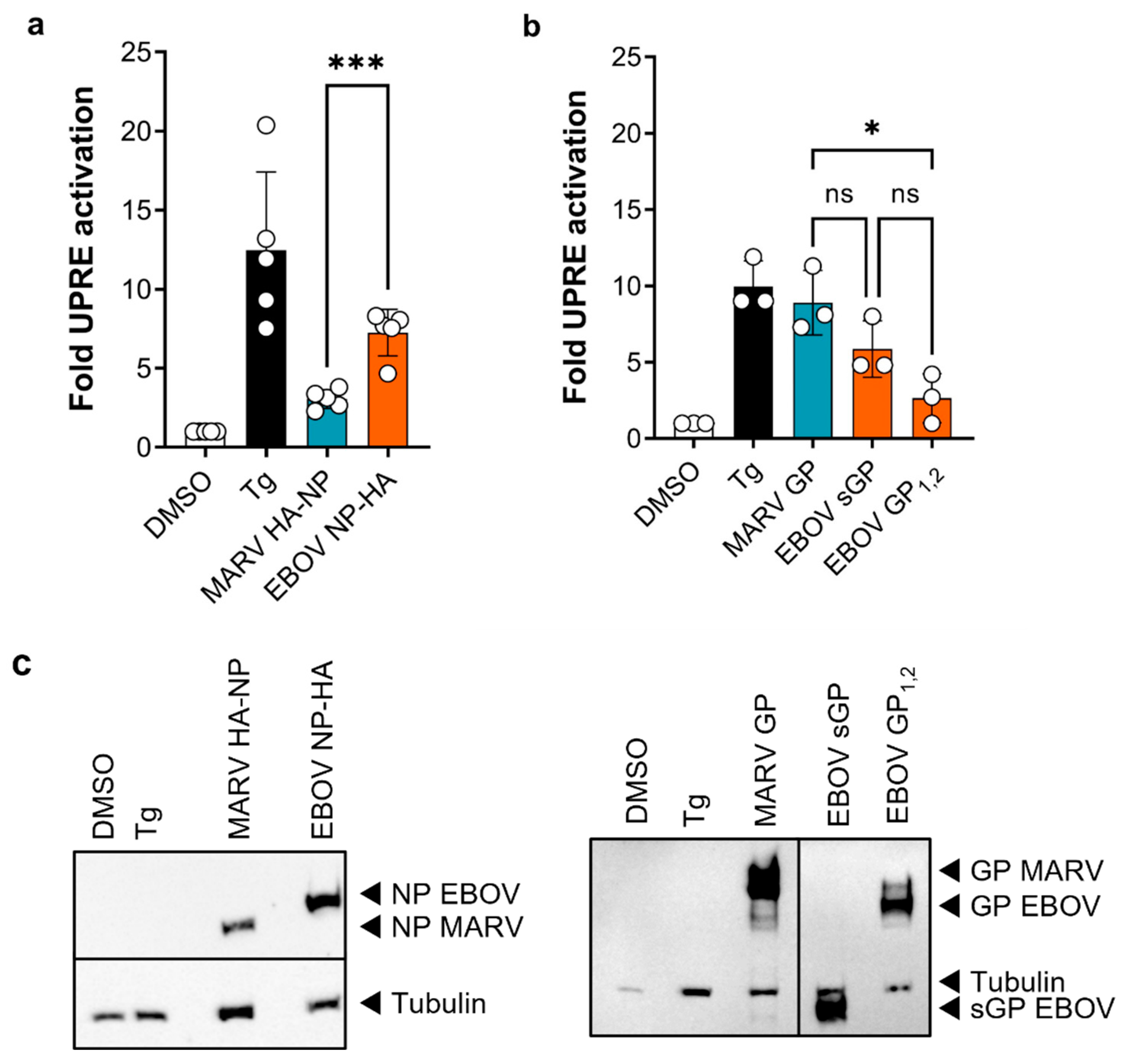
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Korennykh, A.; Walter, P. Structural basis of the unfolded protein response. Annu. Rev. Cell Dev. Biol. 2012, 28, 251–277. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.-H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef] [Green Version]
- Takayanagi, S.; Fukuda, R.; Takeuchi, Y.; Tsukada, S.; Yoshida, K. Gene regulatory network of unfolded protein response genes in endoplasmic reticulum stress. Cell Stress Chaperones 2013, 18, 11–23. [Google Scholar] [CrossRef] [Green Version]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, L. Stress-independent activation of XBP1s and/or ATF6 Rev.eals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef] [Green Version]
- Brunner, J.-M.; Plattet, P.; Doucey, M.-A.; Rosso, L.; Curie, T.; Montagner, A.; Wittek, R.; Vandelvelde, M.; Zurbriggen, A.; Hirling, H.; et al. Morbillivirus glycoprotein expression induces ER stress, alters Ca2+ homeostasis and results in the release of vasostatin. PLoS ONE 2012, 7, e32803. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.-P.; Siu, K.-L.; Chin, K.-T.; Yuen, K.-Y.; Zheng, B.; Jin, D.-Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287. [Google Scholar] [CrossRef] [Green Version]
- Hassan, I.H.; Zhang, M.S.; Powers, L.S.; Shao, J.Q.; Baltrusaitis, J.; Rutkowski, D.T.; Legge, K.; Monick, M.M. Influenza A viral replication is blocked by inhibition of the inositol-requiring enzyme 1 (IRE1) stress pathway. J. Biol. Chem. 2012, 287, 4679–4689. [Google Scholar] [CrossRef] [Green Version]
- Prasad, V.; Suomalainen, M.; Jasiqi, Y.; Hemmi, S.; Hearing, P.; Hosie, L.; Burgert, H.-G.; Greber, U.F. The UPR sensor IRE1α and the adenovirus E3-19K glycoprotein sustain persistent and lytic infections. Nat. Commun. 2020, 11, 1997. [Google Scholar] [CrossRef] [PubMed]
- Rohde, C.; Becker, S.; Krähling, V. Marburg virus regulates the IRE1/XBP1-dependent unfolded protein response to ensure efficient viral replication. Emerg. Microbes. Infect. 2019, 8, 1300–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeDiego, M.L.; Nieto-Torres, J.L.; Jiménez-Guardeño, J.M.; Regla-Nava, J.A.; Alvarez, E.; Oliveros, J.C.; Zhao, J.; Fett, C.; Perlman, S.; Enjuanes, L. Severe acute respiratory syndrome coronavirus envelope protein regulates cell stress response and apoptosis. PLoS Pathog. 2011, 7, e1002315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thastrup, O.; Cullen, P.J.; Drøbak, B.K.; Hanley, M.R.; Dawson, A.P. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase. Proc. Natl. Acad. Sci. USA 1990, 87, 2466–2470. [Google Scholar] [CrossRef] [Green Version]
- Shaban, M.S.; Mayr-Buro, C.; Meier-Soelch, J.; Albert, B.V.; Schmitz, M.L.; Ziebuhr, J.; Kracht, M. Thapsigargin, Key to new host-directed coronavirus antivirals? Trends Pharmacol. Sci. 2022, 43, 557–568. [Google Scholar] [CrossRef] [PubMed]
- Shaban, M.S.; Müller, C.; Mayr-Buro, C.; Weiser, H.; Meier-Soelch, J.; Albert, B.V.; Weber, A.; Linne, U.; Hain, T.; Babayev, I.; et al. Multi-level inhibition of coronavirus replication by chemical ER stress. Nat. Commun. 2021, 12, 5536. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, G.K.; Ayllón, M.A.; Bào, Y.; Basler, C.F.; Bavari, S.; Blasdell, K.R.; Briese, T.; Brown, P.A.; Bukreyev, A.; Balkema-Buschmann, A.; et al. Taxonomy of the order Mononegavirales, Update 2019. Arch. Virol. 2019, 164, 1967–1980. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, M.J. WHO’s Blueprint List of Priority Diseases. JAMA 2018, 319, 1973. [Google Scholar] [CrossRef] [Green Version]
- Emanuel, J.; Marzi, A.; Feldmann, H. Filoviruses, Ecology, Molecular Biology, and Evolution. Adv. Virus Res. 2018, 100, 189–221. [Google Scholar] [CrossRef]
- Messaoudi, I.; Amarasinghe, G.K.; Basler, C.F. Filovirus pathogenesis and immune evasion, Insights from Ebola virus and Marburg virus. Nat. Rev. Microbiol. 2015, 13, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Watanabe, S.; Halfmann, P.; Kawaoka, Y. The spatio-temporal distribution dynamics of Ebola virus proteins and RNA in infected cells. Sci. Rep. 2013, 3, 1206. [Google Scholar] [CrossRef]
- Hoenen, T.; Shabman, R.S.; Groseth, A.; Herwig, A.; Weber, M.; Schudt, G.; Dolnik, O.; Basler, C.F.; Becker, S.; Feldmann, H. Inclusion bodies are a site of ebolavirus replication. J. Virol. 2012, 86, 11779–11788. [Google Scholar] [CrossRef] [Green Version]
- Takamatsu, Y.; Kolesnikova, L.; Becker, S. Ebola virus proteins NP, VP35, and VP24 are essential and sufficient to mediate nucleocapsid transport. Proc. Natl. Acad. Sci. USA 2018, 115, 1075–1080. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Ohba, Y. Budding of Ebola Virus Particles Requires the Rab11-Dependent Endocytic Recycling Pathway. J. Infect. Dis. 2018, 218, S388–S396. [Google Scholar] [CrossRef] [Green Version]
- Schudt, G.; Dolnik, O.; Kolesnikova, L.; Biedenkopf, N.; Herwig, A.; Becker, S. Transport of Ebolavirus Nucleocapsids Is Dependent on Actin Polymerization, Live-Cell Imaging Analysis of Ebolavirus-Infected Cells. J. Infect. Dis. 2015, 212 (Suppl. S2), S160–S166. [Google Scholar] [CrossRef] [PubMed]
- Mittler, E.; Kolesnikova, L.; Herwig, A.; Dolnik, O.; Becker, S. Assembly of the Marburg virus envelope. Cell Microbiol. 2013, 15, 270–284. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Volchkova, V.A.; Ströher, U.; Becker, S.; Dolnik, O.; Cieplik, M.; Garten, W.; Klenk, H.D.; Feldmann, H. Proteolytic processing of Marburg virus glycoprotein. Virology 2000, 268, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Gordon, T.B.; Hayward, J.A.; Marsh, G.A.; Baker, M.L.; Tachedjian, G. Host and Viral Proteins Modulating Ebola and Marburg Virus Egress. Viruses 2019, 11, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.E.; Saphire, E.O. Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 2009, 4, 621–635. [Google Scholar] [CrossRef] [Green Version]
- Mehedi, M.; Falzarano, D.; Seebach, J.; Hu, X.; Carpenter, M.S.; Schnittler, H.-J.; Feldmann, H. A new Ebola virus nonstructural glycoprotein expressed through RNA editing. J. Virol. 2011, 85, 5406–5414. [Google Scholar] [CrossRef] [Green Version]
- Jeffers, S.A.; Sanders, D.A.; Sanchez, A. Covalent modifications of the ebola virus glycoprotein. J. Virol. 2002, 76, 12463–12472. [Google Scholar] [CrossRef]
- Lennemann, N.J.; Walkner, M.; Berkebile, A.R.; Patel, N.; Maury, W. The Role of Conserved N-Linked Glycans on Ebola Virus Glycoprotein 2. J. Infect. Dis. 2015, 212 (Suppl. S2), S204–S209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iraqi, M.; Edri, A.; Greenshpan, Y.; Kundu, K.; Bolel, P.; Cahana, A.; Ottolenghi, A.; Gazit, R.; Lobel, L.; Braiman, A.; et al. N-Glycans Mediate the Ebola Virus-GP1 Shielding of Ligands to Immune Receptors and Immune Evasion. Front. Cell Infect. Microbiol. 2020, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Rayaprolu, V.; Parvate, A.D.; Pronker, M.F.; Hui, S.; Parekh, D.; Shaffer, K.; Yu, X.; Saphire, E.O.; Snijder, J. Glycan shield of the ebolavirus envelope glycoprotein GP. Commun. Biol. 2022, 5, 785. [Google Scholar] [CrossRef] [PubMed]
- Volchkov, V.E.; Feldmann, H.; Volchkova, V.A.; Klenk, H.D. Processing of the Ebola virus glycoprotein by the proprotein convertase furin. Proc. Natl. Acad. Sci. USA 1998, 95, 5762–5767. [Google Scholar] [CrossRef] [Green Version]
- Volchkova, V.A.; Klenk, H.D.; Volchkov, V.E. Delta-peptide is the carboxy-terminal cleavage fragment of the nonstructural small glycoprotein sGP of Ebola virus. Virology 1999, 265, 164–171. [Google Scholar] [CrossRef] [Green Version]
- Ito, H.; Watanabe, S.; Takada, A.; Kawaoka, Y. Ebola virus glycoprotein, Proteolytic processing, acylation, cell tropism, and detection of neutralizing antibodies. J. Virol. 2001, 75, 1576–1580. [Google Scholar] [CrossRef] [Green Version]
- Furuyama, W.; Shifflett, K.; Feldmann, H.; Marzi, A. The Ebola virus soluble glycoprotein contributes to viral pathogenesis by activating the MAP kinase signaling pathway. PLoS Pathog. 2021, 17, e1009937. [Google Scholar] [CrossRef]
- Olejnik, J.; Ryabchikova, E.; Corley, R.B.; Mühlberger, E. Intracellular events and cell fate in filovirus infection. Viruses 2011, 3, 1501–1531. [Google Scholar] [CrossRef] [Green Version]
- Krähling, V.; Becker, D.; Rohde, C.; Eickmann, M.; Eroğlu, Y.; Herwig, A.; Kerber, R.; Kowalski, K.; Vergara-Alert, J.; Becker, S. Development of an antibody capture ELISA using inactivated Ebola Zaire Makona virus. Med. Microbiol. Immunol. 2016, 205, 173–183. [Google Scholar] [CrossRef]
- Koehler, A.; Kolesnikova, L.; Welzel, U.; Schudt, G.; Herwig, A.; Becker, S. A Single Amino Acid Change in the Marburg Virus Matrix Protein VP40 Provides a Replicative Advantage in a Species-Specific Manner. J. Virol. 2016, 90, 1444–1454. [Google Scholar] [CrossRef]
- Pauly, D.; Chacana, P.A.; Calzado, E.G.; Brembs, B.; Schade, R. IgY technology, Extraction of chicken antibodies from egg yolk by polyethylene glycol (PEG) precipitation. J. Vis. Exp. 2011, 51, e3084. [Google Scholar] [CrossRef] [PubMed]
- Hoenen, T.; Groseth, A.; Kolesnikova, L.; Theriault, S.; Ebihara, H.; Hartlieb, B.; Bamberg, S.; Feldmann, H.; Ströher, U.; Becker, S. Infection of naive target cells with virus-like particles, Implications for the function of ebola virus VP24. J. Virol. 2006, 80, 7260–7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA binding site by the endoplasmic reticulum stress response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [Green Version]
- Lipson, K.L.; Ghosh, R.; Urano, F. The role of IRE1alpha in the degradation of insulin mRNA in pancreatic beta-cells. PLoS ONE 2008, 3, e1648. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.-M.; Chen, Y.-J.; Ouyang, H.-J. Regulation of unfolded protein response modulator XBP1s by acetylation and deacetylation. Biochem. J. 2011, 433, 245–252. [Google Scholar] [CrossRef] [Green Version]
- Hempstead, A.D.; Isberg, R.R. Inhibition of host cell translation elongation by Legionella pneumophila blocks the host cell unfolded protein response. Proc. Natl. Acad. Sci. USA 2015, 112, E6790–E6797. [Google Scholar] [CrossRef] [Green Version]
- Krähling, V.; Stein, D.A.; Spiegel, M.; Weber, F.; Mühlberger, E. Severe acute respiratory syndrome coronavirus triggers apoptosis via protein kinase R but is resistant to its antiviral activity. J. Virol. 2009, 83, 2298–2309. [Google Scholar] [CrossRef] [Green Version]
- Dolnik, O.; Kolesnikova, L.; Stevermann, L.; Becker, S. Tsg101 is recruited by a late domain of the nucleocapsid protein to support budding of Marburg virus-like particles. J. Virol. 2010, 84, 7847–7856. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lucius, M.D.; Altomare, D.; Havighorst, A.; Farmaki, E.; Chatzistamou, I.; Shtutman, M.; Kiaris, H. Coordination Analysis of Gene Expression Points to the Relative Impact of Different Regulators During Endoplasmic Reticulum Stress. DNA Cell Biol. 2019, 38, 969–981. [Google Scholar] [CrossRef]
- Feldmann, H.; Bugany, H.; Mahner, F.; Klenk, H.D.; Drenckhahn, D.; Schnittler, H.J. Filovirus-induced endothelial leakage triggered by infected monocytes/macrophages. J. Virol. 1996, 70, 2208–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Korennykh, A.V.; Behrman, S.L.; Walter, P. Mammalian endoplasmic reticulum stress sensor IRE1 signals by dynamic clustering. Proc. Natl. Acad. Sci. USA 2010, 107, 16113–16118. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Hope, T.J. Full-length Ebola glycoprotein accumulates in the endoplasmic reticulum. Virol. J. 2011, 8, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.-M.; Kang, T.-I.; So, J.-S. Roles of XBP1s in Transcriptional Regulation of Target Genes. Biomedicines 2021, 9, 791. [Google Scholar] [CrossRef] [PubMed]
- Karagöz, G.E.; Acosta-Alvear, D.; Walter, P. The Unfolded Protein Response, Detecting and Responding to Fluctuations in the Protein-Folding Capacity of the Endoplasmic Reticulum. Cold Spring Harb. Perspect. Biol. 2019, 11, a033886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Conza, G.; Ho, P.-C. ER Stress Responses, An Emerging Modulator for Innate Immunity. Cells 2020, 9, 695. [Google Scholar] [CrossRef] [Green Version]
- Zhu, E.; Chen, W.; Qin, Y.; Ma, S.; Fan, S.; Wu, K.; Li, W.; Fan, J.; Yi, L.; Ding, H.; et al. Classical Swine Fever Virus Infection Induces Endoplasmic Reticulum Stress-Mediated Autophagy to Sustain Viral Replication in vivo and in vitro. Front. Microbiol. 2019, 10, 2545. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Dong, J.; Zhu, S.; Yuan, F.; Wei, L.; Wang, J.; Quan, R.; Chu, J.; Wang, D.; Jiang, H.; et al. Seneca valley virus activates autophagy through the PERK and ATF6 UPR pathways. Virology 2019, 537, 254–263. [Google Scholar] [CrossRef]
- Abdullah, A.; Ravanan, P. The unknown face of IRE1α—Beyond ER stress. Eur. J. Cell Biol. 2018, 97, 359–368. [Google Scholar] [CrossRef]
- Flores-Santibáñez, F.; Medel, B.; Bernales, J.I.; Osorio, F. Understanding the Role of the Unfolded Protein Response Sensor IRE1 in the Biology of Antigen Presenting Cells. Cells 2019, 8, 1563. [Google Scholar] [CrossRef]
- Urra, H.; Pihán, P.; Hetz, C. The UPRosome—Decoding novel biological outputs of IRE1α function. J. Cell Sci. 2020, 133, jcs218107. [Google Scholar] [CrossRef]
- Drori, A.; Messerle, M.; Brune, W.; Tirosh, B. Lack of XBP-1 impedes murine cytomegalovirus gene expression. PLoS ONE 2014, 9, e110942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stahl, S.; Burkhart, J.M.; Hinte, F.; Tirosh, B.; Mohr, H.; Zahedi, R.P.; Sickmann, A.; Ruzsics, Z.; Budt, M.; Brune, W. Cytomegalovirus downregulates IRE1 to repress the unfolded protein response. PLoS Pathog. 2013, 9, e1003544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, P.; Christensen, S.B.; Nissen, P.; Møller, J.V.; Engedal, N. Cell death induced by the ER stressor thapsigargin involves death receptor 5, a non-autophagic function of MAP1LC3B, and distinct contributions from unfolded protein response components. Cell Commun. Signal. 2020, 18, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Beltagi, S.; Preda, C.A.; Goulding, L.V.; James, J.; Pu, J.; Skinner, P.; Jiang, Z.; Wang, B.L.; Yang, J.; Banyard, A.C.; et al. Thapsigargin Is a Broad-Spectrum Inhibitor of Major Human Respiratory Viruses, Coronavirus, Respiratory Syncytial Virus and Influenza A Virus. Viruses 2021, 13, 234. [Google Scholar] [CrossRef]
- Kumar, N.; Khandelwal, N.; Kumar, R.; Chander, Y.; Rawat, K.D.; Chaubey, K.K.; Sharma, S.; Singh, S.V.; Riyesh, T.; Tripathi, B.N.; et al. Inhibitor of Sarco/Endoplasmic Reticulum Calcium-ATPase Impairs Multiple Steps of Paramyxovirus Replication. Front. Microbiol. 2019, 10, 209. [Google Scholar] [CrossRef]
- Yura, Y.; Matsumoto, R.; Sumi, T.; Kusaka, J. Effect of Ca2+-dependent cell death on the release of herpes simplex virus. Arch. Virol. 2003, 148, 221–235. [Google Scholar] [CrossRef]
- Taverner, W.K.; Jacobus, E.J.; Christianson, J.; Champion, B.; Paton, A.W.; Paton, J.C.; Su, W.; Cawood, R.; Seymour, L.W.; Lei-Rossmann, J. Calcium Influx Caused by ER Stress Inducers Enhances Oncolytic Adenovirus Enadenotucirev Replication and Killing through PKCα Activation. Mol. Ther. Oncolytics 2019, 15, 117–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, S.P.; Shurtleff, A.C.; Costantino, J.A.; Tritsch, S.R.; Retterer, C.; Spurgers, K.B.; Bavari, S. HSPA5 is an essential host factor for Ebola virus infection. Antiviral. Res. 2014, 109, 171–174. [Google Scholar] [CrossRef]
- Kotliar, D.; Lin, A.E.; Logue, J.; Hughes, T.K.; Khoury, N.M.; Raju, S.S.; Wadsworth, M.H.; Chen, H.; Kurtz, J.R.; Dighero-Kemp, B.; et al. Single-Cell Profiling of Ebola Virus Disease In Vivo Reveals Viral and Host Dynamics. Cell 2020, 183, 1383–1401. [Google Scholar] [CrossRef]
- Volchkov, V.E.; Chepurnov, A.A.; Volchkova, V.A.; Ternovoj, V.A.; Klenk, H.D. Molecular characterization of guinea pig-adapted variants of Ebola virus. Virology 2000, 277, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volchkova, V.A.; Dolnik, O.; Martinez, M.J.; Reynard, O.; Volchkov, V.E. RNA Editing of the GP Gene of Ebola Virus is an Important Pathogenicity Factor. J. Infect. Dis. 2015, 212 (Suppl. S2), S226–S233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosworth, A.; Dowall, S.D.; Armstrong, S.; Liu, X.; Dong, X.; Bruce, C.B.; F P Ng, L.; Carroll, M.W.; Hewson, R.; Hiscox, J.A. Investigating the Cellular Transcriptomic Response Induced by the Makona Variant of Ebola Virus in Differentiated THP-1 Cells. Viruses 2019, 11, 1023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.H.; Lee, K.; Vink, E.; Kaufman, R.J. Cytoplasmic IRE1alpha-mediated XBP1 mRNA splicing in the absence of nuclear processing and endoplasmic reticulum stress. J. Biol. Chem. 2006, 281, 18691–18706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, T.A.; Junjappa, R.P.; Handigund, M.; Ferdous, J.; Kim, H.-R.; Chae, H.-J. Role of Endoplasmic Reticulum Stress Sensor IRE1α in Cellular Physiology, Calcium, ROS Signaling, and Metaflammation. Cells 2020, 9, 1160. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, J.; Liu, X.; Chai, Q.; Lu, X.; Yao, X.; Yang, Z.; Sun, L.; Johnson, S.F.; Schwartz, R.C.; et al. Protein disulfide isomerases (PDIs) negatively regulate ebolavirus structural glycoprotein expression in the endoplasmic reticulum (ER) via the autophagy-lysosomal pathway. Autophagy 2022, 18, 2350–2367. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, B.; Gao, X.; Peng, C.; Shan, C.; Johnson, S.F.; Schwartz, R.C.; Zheng, Y.-H. RNF185 regulates proteostasis in Ebolavirus infection by crosstalk between the calnexin cycle, ERAD, and reticulophagy. Nat. Commun. 2022, 13, 6007. [Google Scholar] [CrossRef]
- Page, A.; Volchkova, V.A.; Reid, S.P.; Mateo, M.; Bagnaud-Baule, A.; Nemirov, K.; Shurtleff, A.C.; Lawrence, P.; Reynard, O.; Ottmann, M.; et al. Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell Rep. 2014, 6, 1026–1036. [Google Scholar] [CrossRef]
- Edwards, M.R.; Johnson, B.; Mire, C.E.; Xu, W.; Shabman, R.S.; Speller, L.N.; Leung, D.W.; Geisbert, T.W.; Amarasinghe, G.K.; Basler, C.F. The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep. 2014, 6, 1017–1025. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rohde, C.; Pfeiffer, S.; Baumgart, S.; Becker, S.; Krähling, V. Ebola Virus Activates IRE1α-Dependent XBP1u Splicing. Viruses 2023, 15, 122. https://doi.org/10.3390/v15010122
Rohde C, Pfeiffer S, Baumgart S, Becker S, Krähling V. Ebola Virus Activates IRE1α-Dependent XBP1u Splicing. Viruses. 2023; 15(1):122. https://doi.org/10.3390/v15010122
Chicago/Turabian StyleRohde, Cornelius, Sebastian Pfeiffer, Sara Baumgart, Stephan Becker, and Verena Krähling. 2023. "Ebola Virus Activates IRE1α-Dependent XBP1u Splicing" Viruses 15, no. 1: 122. https://doi.org/10.3390/v15010122