
Predicting Epitope Candidates for SARS-CoV-2

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Description of Data Used
2.1.1. Immune Epitope Database
- 1.
- Epitope ID;
- 2.
- Parent Protein;
- 3.
- Parent Protein Accession;
- 4.
- Antigen Name;
- 5.
- Epitope Description (Sequence).
2.1.2. SARS-CoV-2 Genomic Data
2.2. Protein Sequence Diversity Analysis
2.3. Identification of Conserved Epitope Sequences
- Identification of whether an epitope occurs on a protein sequence and at what locus (amino acid position) using exact string matching functions provided in base Python. For this, the epitope’s parent protein was matched to the SARS-CoV-2 protein, e.g., epitopeA, whose parent protein indicated in IEDB is Nucleoprotein, would be checked for presence on SARS-CoV-2 Nucleoprotein sequences only;
- Calculation of the number of times an epitope occurs on a protein sequence.
2.4. Identification of Candidate Epitope Sequences
2.5. Identification of T Cell MHC Class I Epitopes
2.6. Summary Statistics
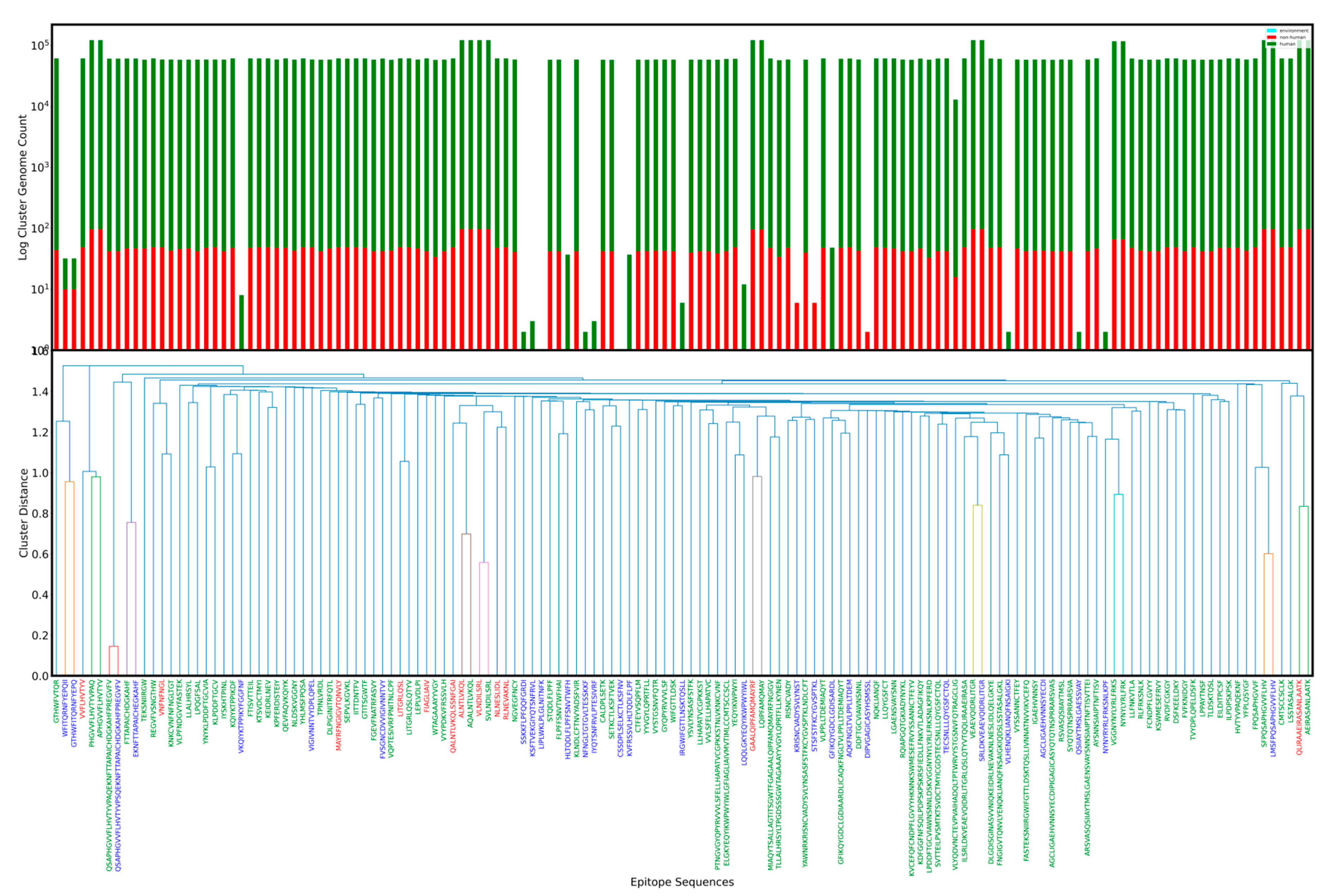
2.7. Epitope Clustering and Classes
- 1.
- Epitopes (both ORIGINAL and CANDIDATE) were grouped based on functional type (T or B) and parent protein;
- 2.
- For each group above, the Levenshtein edit distance measure was calculated (using Python) for every epitope–epitope pair. This yields a square matrix with axes labeled by epitope and cell values as the distances between all pairs;
- 3.
- Linkage (Python scipy package) was run on the edit distance matrix using ’Euclidean’ metric and ’Single’ method;
- 4.
- The resulting linkage matrix was used to compute and plot a dendrogram;
- 5.
- A cluster threshold was defined where cluster members had a length normalized edit distance less than one. We color any linkage lineage lines not clustering together in blue.
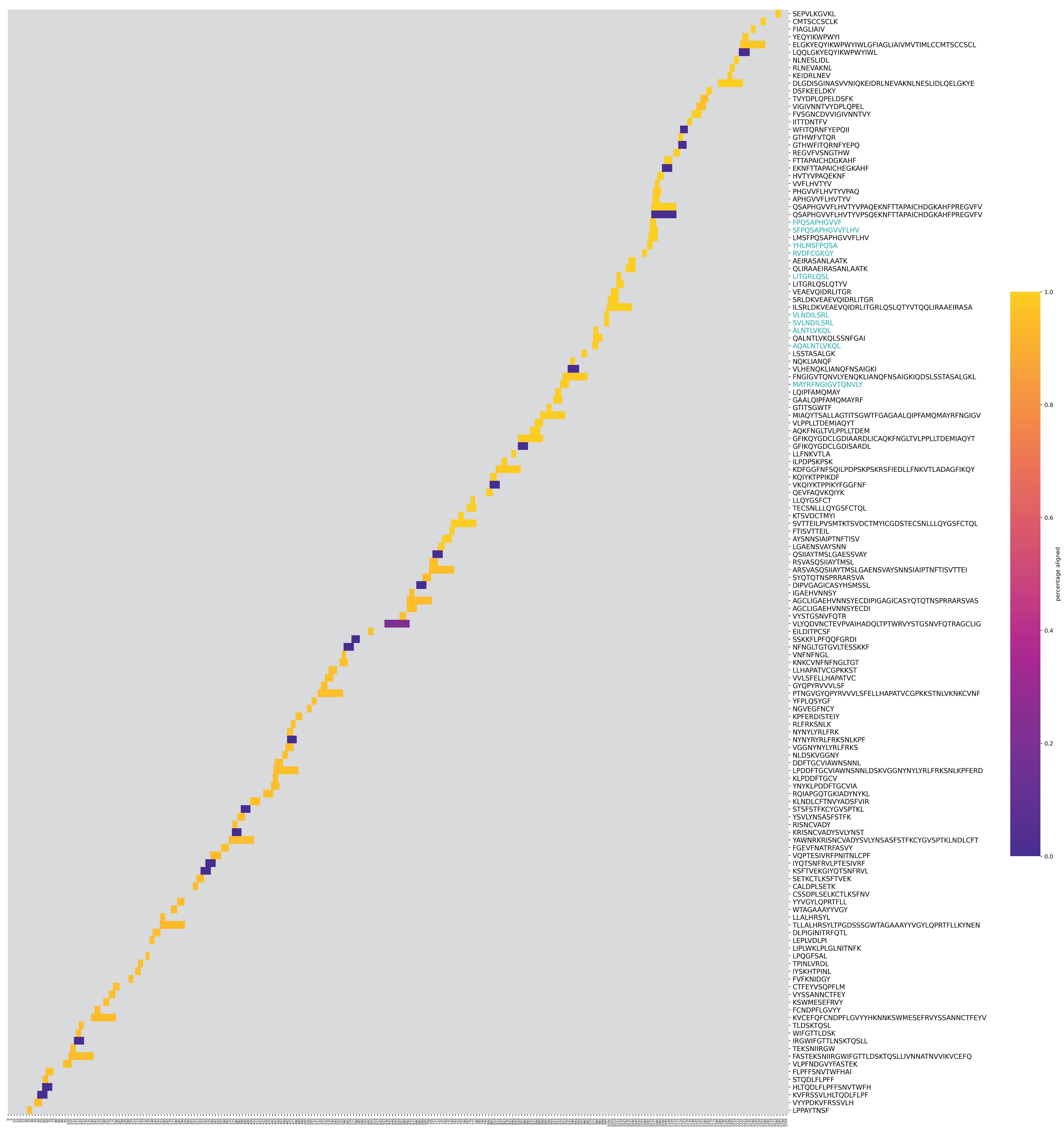
- 1.
- B cell and T cell epitopes were divided into separate sets based on parent protein as described above;
- 2.
- To remove spurious sequences or assembly errors, only sequences with length within of the UniProt [54] reference protein sequence for SARS-CoV-2 were considered, e.g., for Nucleoprotein, the length of reference protein is 419 amino acids, and all sequences that were within a ±10% range of that, i.e., between 378 and 461 amino acids in length, were allowed. For SARS-CoV-2 Spike glycoprotein, the reference protein is of length 1273 amino acids and the allowed range was 1146 to 1400 amino acid characters;
- 3.
- Furthermore, multi-sequence alignments (MSA) were run for all sequences per protein name relative to one another using MAFFT (v7.431) with the reorder option [55];
- 4.
- For each protein, a matrix was then constructed, with the x-axis indicating the amino acid position on the protein and the y-axis indicating the epitopes where the length of the x-axis was the maximum length of the protein sequence;
- 5.
- Each cell was then assigned a binary value, i.e., 0 or 1. The cell is assigned 1 if the epitope on that row is found on the position on the protein corresponding with the column index of the cell, e.g., if ‘epitopeA’ is on row 0 and is found on a Nucleoprotein sequence between indices 7 and 15, then index 7 would be filled with 1;
- 6.
- After sequences have been aligned in the MSA, the positional information was padded so that all pairwise comparisons are represented with identical start and stop coordinates, and gaps are filled in with dashes. To accommodate this relative positional information generated by an MSA, regular expressions were used to identify the start index of epitope sequences with the allowance for epitopes that span “gapped regions” due to the filling in of sequences in the MSA;
- 7.
- Epitope sequences spanning regions of insertion described in step 6 were investigated further for key non-synonymous mutations and their prevalence in this corpus of ORIGINAL and CANDIDATE epitopes;
- 8.
- After alignment, the epitopes were identified on the same position within the protein sequences. The bars representing epitopes were colored by their presence across the genomes;
- 9.
- After constructing the matrix in steps 2 and 3, ‘single’ linkage was run using method ‘Euclidean’ metric;
- 10.
- Finally, the clustermap and dendrogram were plotted using Python’s seaborn library.
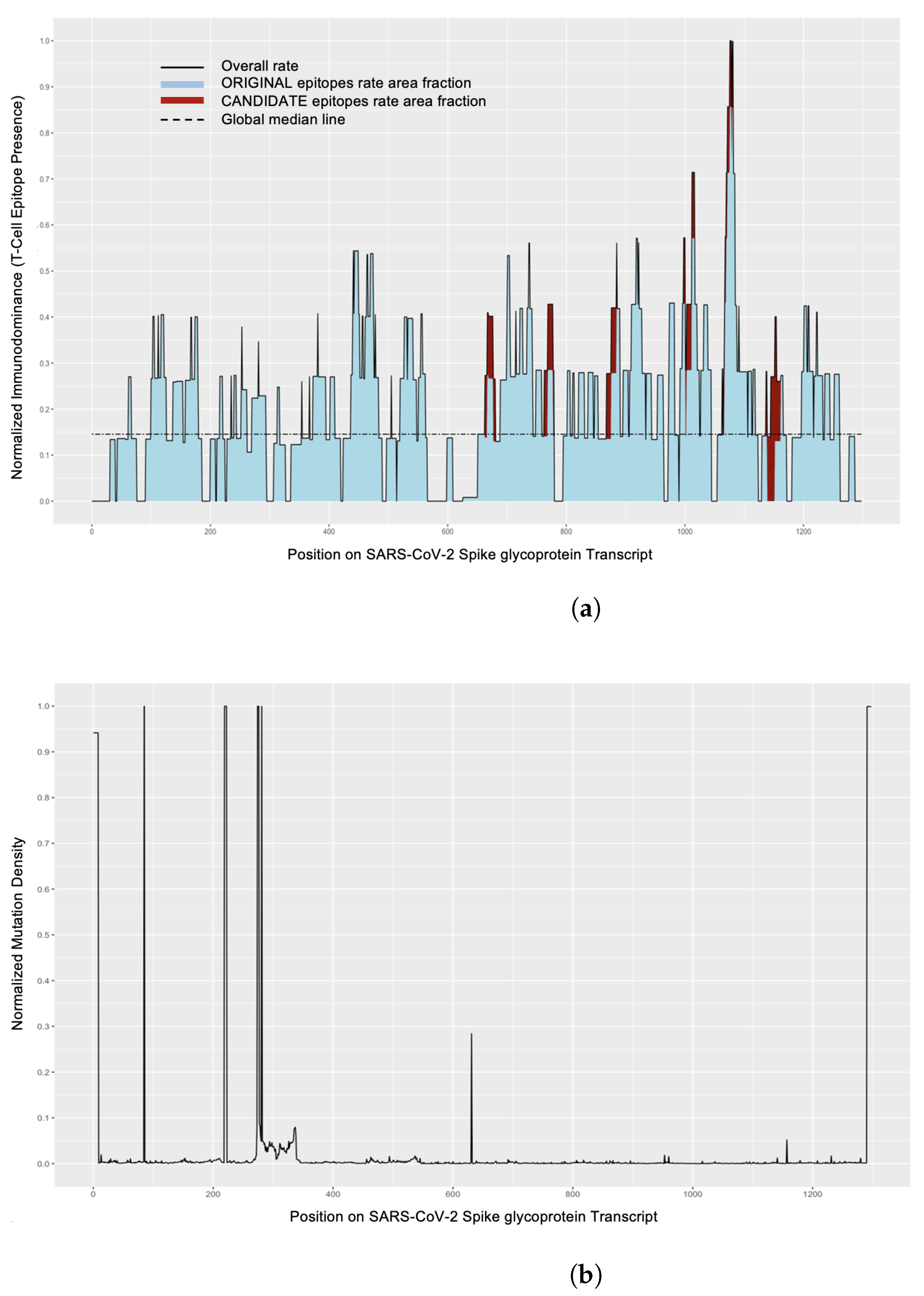
2.8. Identification of Epitopes on Protein
2.8.1. Mutations in Proteins Affecting Epitopes and Mutation Density
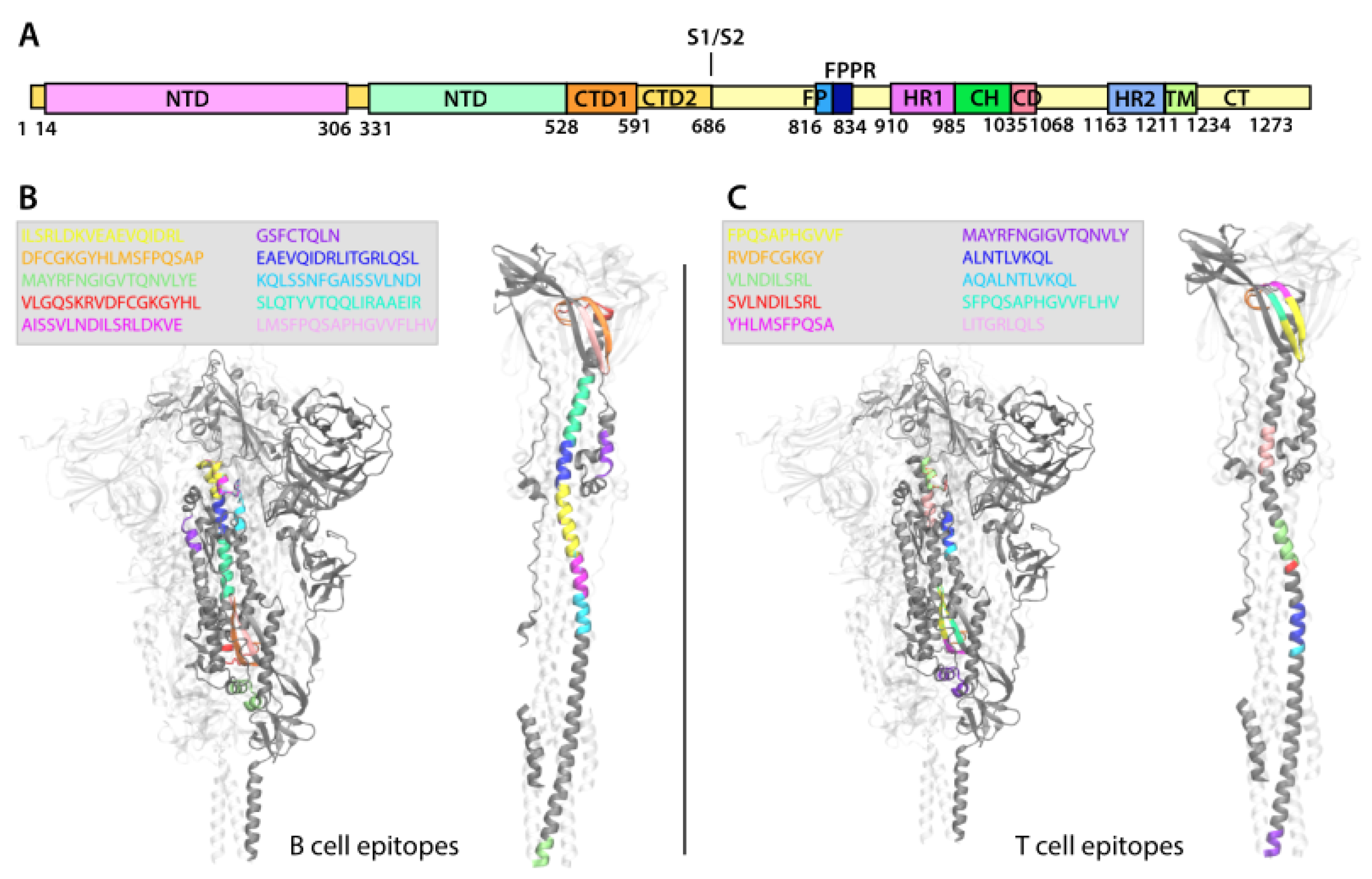
2.8.2. Epitope Distribution across Protein
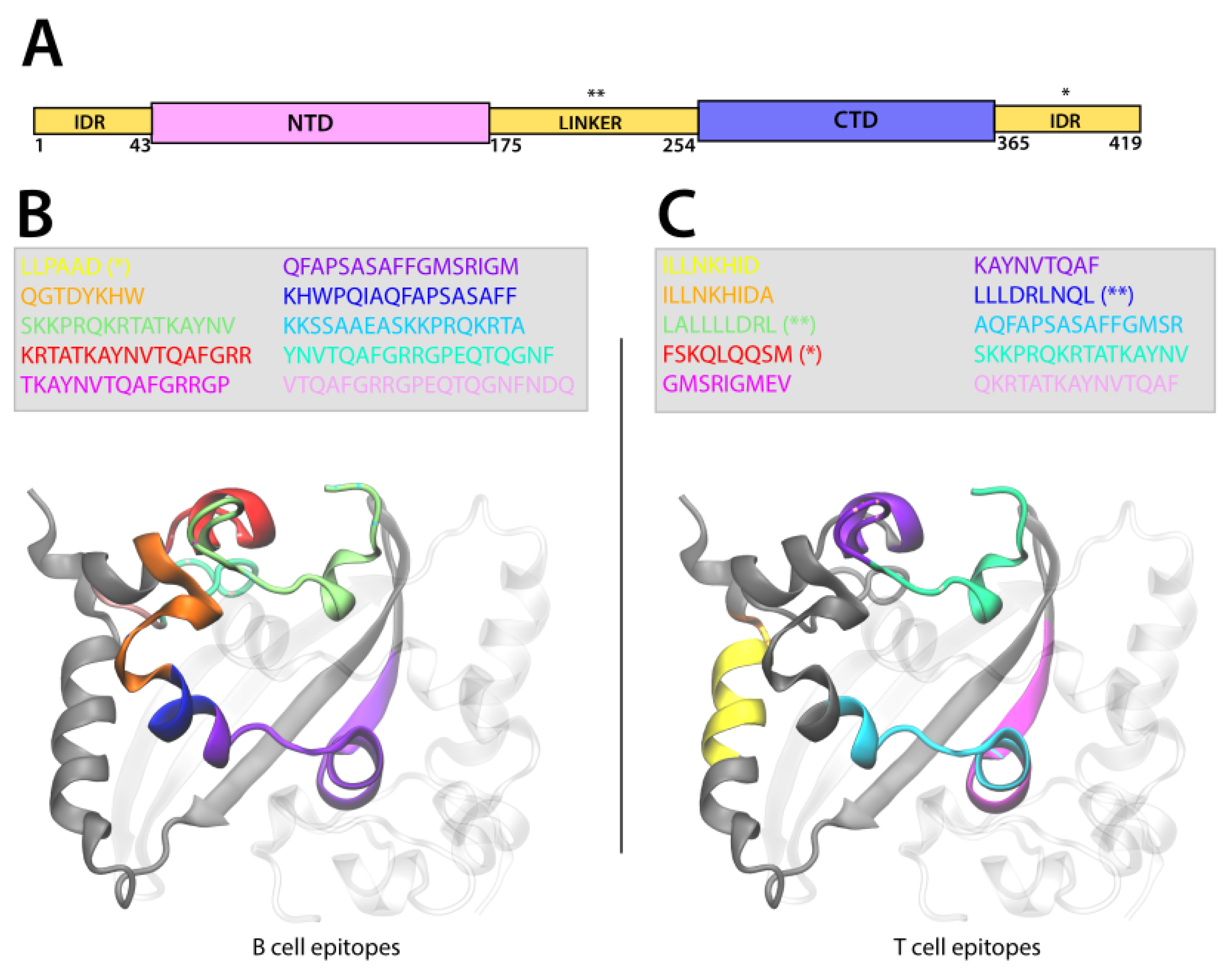
2.8.3. Epitope Localization on Protein Structure
2.9. Result Verification
3. Results
3.1. Protein Sequence Diversity Analysis
3.2. Epitope Sequences
3.3. T Cell MHC Class I Peptides
3.4. Epitopes on Protein
3.4.1. Mutations in Proteins Affecting Epitopes
3.4.2. Epitope Distribution across Protein
3.4.3. Epitope Localization on Protein Structure
4. Discussion
- 1.
- From sequence based clustering (Figure 1), it is possible measure the evolution rate for a cluster by analyzing the ratio of candidate to total epitopes in a cluster. This metric can be useful when evaluating epitope candidates for vaccines and can be used to theoretically predict the probability of change of an epitope solely on sequence homology. Additionally, mapping cluster presence across a genome set adds a dimension for identifying the most suitable epitopes. Additionally, it is possible to filter by host to study changes that might arise from a virus evolving in different hosts. Tracking of major clusters could also enable development of statistical models to estimate a timeline for immune response robustness.
- 2.
- By analyzing mutation density regions and immunodominance regions, it is possible to evaluate which segments of proteins may be undergoing the fewest amino acid changes and thus would advise the most stable regions on the protein or those that may be evolving under selective pressure. This type of analysis combined with studying position-based clustering could provide more insight for selection of epitopes, as it would highlight most prevalent epitopes with consideration for their neighbors.
5. Future Work
- 1.
- It is possible to study the proximity of epitopes to more mutation prone regions to eliminate immune targets that otherwise may seem promising.
- 2.
- It is possible to also combine the epitope evaluation approaches discussed in Section 4 to design a quantitative metric for evaluating and ranking epitope targets suitable for vaccine development. Selection of stable epitopes is important, as that would enable development of shorter vaccines and may have a positive impact on effectiveness of vaccines because more stable immunotargets may be learned by the host immune system.
- 3.
- It would be interesting scientifically to study the presence of the top ten epitopes in vaccine sequences.
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
- IBM Functional Genomics Platform (ibm.biz/functional-genomics);
- Publication regarding FGP’s annotation of SARS-CoV-2 data [46];
- Delta S sequences are in Supplemental Data SD1;
- Reference sequences for N and S are in Supplemental Data SD8.
Conflicts of Interest
References
- Sanchez-Trincado, J.L.; Gomez-Perosanz, M.; Reche, P.A. Fundamentals and methods for T-and B-cell epitope prediction. J. Immunol. Res. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, M.; Abualrous, E.T.; Sticht, J.; Álvaro Benito, M.; Stolzenberg, S.; Noé, F.; Freund, C. Major histocompatibility complex (MHC) class I and MHC class II proteins: Conformational plasticity in antigen presentation. Front. Immunol. 2017, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Prachar, M.; Justesen, S.; Steen-Jensen, D.B.; Thorgrimsen, S.; Jurgons, E.; Winther, O.; Bagger, F.O. Identification and validation of 174 COVID-19 vaccine candidate epitopes reveals low performance of common epitope prediction tools. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ting, S.; Yufei, H.; Wendong, L.; Yubo, F.; Jing, Z. Epitope-based peptide vaccines predicted against novel coronavirus disease caused by SARS-CoV-2. Virus Res. 2020, 288, 198082. [Google Scholar] [CrossRef] [PubMed]
- Bahai, A.; Asgari, E.; Mofrad, M.R.K.; Kloetgen, A.; McHardy, A.C. EpitopeVec: Linear epitope prediction using deep protein sequence embeddings. Bioinformatics 2021, 37, 4517–4525. [Google Scholar] [CrossRef] [PubMed]
- Jespersen, M.C.; Peters, B.; Nielsen, M.; Marcatili, P. BepiPred-2.0: Improving sequence-based B-cell epitope prediction using conformational epitopes. Nucleic Acids Res. 2017, 45, W24–W29. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Sidney, J.; Zhang, Y.; Scheuermann, R.H.; Peters, B.; Sette, A. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 2020, 27, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.S.; Filho, V.B.; Calomeno, N.A.; Wagner, G.; Miletti, L.C. EpiBuilder: A Tool for Assembling, Searching, and Classifying B-Cell Epitopes. Bioinform. Biol. Insights 2022, 16, 11779322221095221. [Google Scholar] [CrossRef]
- Saha, S.; Raghava, G.P.S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins Struct. Funct. Bioinform. 2006, 65, 40–48. [Google Scholar] [CrossRef]
- Yao, B.; Zhang, L.; Liang, S.; Zhang, C. SVMTriP: A Method to Predict Antigenic Epitopes Using Support Vector Machine to Integrate Tri-Peptide Similarity and Propensity. PLoS ONE 2012, 7, e45152. [Google Scholar] [CrossRef] [PubMed]
- Collatz, M.; Mock, F.; Barth, E.; Hölzer, M.; Sachse, K.; Marz, M. EpiDope: A deep neural network for linear B-cell epitope prediction. Bioinformatics 2020, 37, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Andreatta, M.; Nielsen, M. Gapped sequence alignment using artificial neural networks: Application to the MHC class I system. Bioinformatics 2016, 32, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Du, N.; Lei, Y.; Dorje, S.; Qi, J.; Luo, T.; Gao, G.F.; Song, H. Structures of the SARS-CoV-2 nucleocapsid and their perspectives for drug design. EMBO J. 2020, 39, e105938. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Pan, J.; Tao, J.; Guo, D. SARS-CoV nucleocapsid protein antagonizes IFN-b response by targeting initial step of IFN-b induction pathway, and its C-terminal region is critical for the antagonism. Virus Genes 2011, 42, 37–45. [Google Scholar] [CrossRef]
- Duan, L.; Zheng, Q.; Zhang, H.; Niu, Y.; Lou, Y.; Wang, H. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: Implications for the design of spike-based vaccine immunogens. Front. Immunol. 2020, 576622. [Google Scholar] [CrossRef] [PubMed]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Zuniga, S.; Cruz, J.L.; Sola, I.; Mateos-Gomez, P.A.; Palacio, L.; Enjuanes, L. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 2010, 84, 2169–2175. [Google Scholar] [CrossRef] [PubMed]
- Cong, Y.; Ulasli, M.; Schepers, H.; Mauthe, M.; V’Kovski, P.; Kriegenburg, F.; Thiel, V.; de Haan, C.A.M.; Reggiori, F. Nucleocapsid protein recruitment to replication-transcription complexes plays a crucial role in coronaviral life cycle. J. Virol. 2020, 94, e01925-19. [Google Scholar] [CrossRef]
- Surjit, M.; Liu, B.; Jameel, S.; Chow, V.T.; Lal, S.K. The SARS coronavirus nucleocapsid protein induces actin reorganization and apoptosis in COS-1 cells in the absence of growth factors. Biochem. J. 2004, 383, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Surjit, M.; Liu, B.; Chow, V.T.; Lal, S.K. The nucleocapsid protein of severe acute respiratory syndrome-coronavirus inhibits the activity of cyclin-cyclin-dependent kinase complex and blocks S phase progression in mammalian cells. J. Biol. Chem. 2006, 281, 10669–10681. [Google Scholar] [CrossRef]
- Steuler, H.; Schröder, B.; Bürger, H.; Scholtissek, C. Sequence of the nucleoprotein gene of influenza A/parrot/Ulster/73. Virus Res. 1985, 3, 35–40. [Google Scholar] [CrossRef]
- Chang, C.K.; Hou, M.H.; Chang, C.F.; Hsiao, C.D.; Huang, T.H. The SARS coronavirus nucleocapsid protein–forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Chang, C.K.; Chang, Y.W.; Sue, S.C.; Bai, H.I.; Riang, L.; Hsiao, C.D.; Huang, T.H. Structure of the SARS coronavirus nucleocapsid protein RNA-binding dimerization domain suggests a mechanism for helical packaging of viral RNA. J. Mol. Biol. 2007, 368, 1075–1086. [Google Scholar] [CrossRef] [PubMed]
- Saikatendu, K.S.; Joseph, J.S.; Subramanian, V.; Neuman, B.W.; Buchmeier, M.J.; Stevens, R.C.; Kuhn, P. Ribonucleocapsid formation of severe acute respiratory syndrome coronavirus through molecular action of the N-terminal domain of N protein. J. Virol. 2007, 81, 3913–3921. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Liu, C.L.; Chang, Y.M.; Zhao, J.; Perlman, S.; Hou, M.H. Structural basis for the identification of the N-terminal domain of coronavirus nucleocapsid protein as an antiviral target. J. Med. Chem. 2014, 63, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Szelazek, B.; Kabala, W.; Kus, K.; Zdzalik, M.; Twarda-Clapa, A.; Golik, P.; Burmistrz, M.; Florek, D.; Wladyka, B.; Pyrc, K. Structural characterization of human coronavirus NL63 N protein. J. Virol. 2017, 91, e02503. [Google Scholar] [CrossRef]
- Nguyen, T.H.V.; Lichiere, J.; Canard, B.; Papageorgiou, N.; Attoumani, S.; Ferron, F.; Coutard, B. Structure and oligomerization state of the C-terminal region of the Middle East respiratory syndrome coronavirus nucleoprotein. Acta Crystallogr. D Struct. Biol. 2019, 75, 8–15. [Google Scholar] [CrossRef]
- Cubuk, J.; Alston, J.J.; Incicco, J.J.; Singh, S.; Stuchell-Brereton, M.D.; Ward, M.D.; Zimmerman, M.I.; Vithani, N.; Griffith, D.; Wagoner, J.A.; et al. The SARS-CoV-2 nucleocapsid protein is dynamic, disordered, and phase separates with RNA. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Zinzula, L.; Basquin, J.; Bohn, S.; Beck, F.; Klumpe, S.; Pfeifer, G.; Nagy, I.; Bracher, A.; Hartl, F.U.; Baumeister, W. High-resolution structure and biophysical characterization of the nucleocapsid phosphoprotein dimerization domain from the Covid-19 severe acute respiratory syndrome coronavirus 2. Biochem. Biophys. Res. Commun. 2021, 538, 54–62. [Google Scholar] [CrossRef]
- Ye, Q.; West, A.M.V.; Silletti, S.; Corbett, K.D. Architecture and self-assembly of the SARS-CoV-2 nucleocapsid protein. Protein Sci. 2020, 29, 1890–1901. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Yang, M.; Hong, Z.; Zhang, L.; Huanga, Z.; Chen, X.; He, S.; Zhoua, Z.; Zhoua, Z.; Chen, Q.; et al. Crystal structure of SARS-CoV-2 nucleocapsid protein RNA domain reveals potential unique drug targeting sites. Acta Pharm. Sin. B 2020, 10, 1228–1238. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef] [PubMed]
- Li, F. Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Tortorici, M.A.; Snijder, J.; Xiong, X.; Bosch, B.J.; Rey, F.A.; Veesler, D. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc. Natl. Acad. Sci. USA 2017, 114, 11157–11162. [Google Scholar] [CrossRef] [PubMed]
- Duquerroy, S.; Vigouroux, A.; Rottier, P.J.M.; Rey, F.A.; Bosch, B.J. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology 2005, 335, 276–285. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 180, 1–12. [Google Scholar]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J. The coronavirus spike protein is a class I virus fusion protein: Structural and functional characterization of the fusion core complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef]
- Tortorici, M.A.; Veesler, D. Structural insights into coronavirus entry. Adv. Virus Res. 2019, 105, 94–108. [Google Scholar]
- Kielian, M. Mechanisms of virus membrane fusion proteins. Annu. Rev. Virol. 2014, 1, 171–189. [Google Scholar] [CrossRef]
- Harrison, S.C. Viral membrane fusion. Virology 2015, 479, 498–507. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Dessen, A.; Harrison, L.J.C.S.C.; Skehel, J.J.; Wiley, D.C. Structural basis for membrane fusion by enveloped viruses. Mol. Memb. Biol. 1999, 16, 3–9. [Google Scholar] [CrossRef]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [PubMed]
- Seabolt, E.; Nayar, G.; Krishnareddy, H.; Agarwal, A.; Beck, K.L.; Kandogan, E.; Kuntomi, M.; Roth, M.; Terrizzano, I.; Kaufman, J.; et al. IBM Functional Genomics Platform, A Cloud-Based Platform for Studying Microbial Life at Scale. IEEE/ACM Trans. Comput. Biol. Bioinform. 2020, 19, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Beck, K.L.; Seabolt, E.; Agarwal, A.; Nayar, G.; Bianco, S.; Krishnareddy, H.; Ngo, T.A.; Kunitomi, M.; Mukherjee, V.; Kaufman, J.H. Semi-Supervised Pipeline for Autonomous Annotation of SARS-CoV-2 Genomes. Viruses 2021, 13, 2426. [Google Scholar] [CrossRef]
- Benson, D.A.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2009, 37, D26–D31. [Google Scholar] [CrossRef]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Challenges 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Finkel, Y.; Mizrahi, O.; Nachshon, A.; Weingarten-Gabbay, S.; Morgenstern, D.; Yahalom-Ronen, Y.; Tamir, H.; Achdout, H.; Stein, D.; Israeli, O.; et al. The coding capacity of SARS-CoV-2. Nature 2021, 589, 125–130. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Karlin, S.; Altschul, S.F. Methods for assessing the statistical significance of molecular sequence features by using general scoring schemes. Proc. Natl. Acad. Sci. USA 1990, 87, 2264–2268. [Google Scholar] [CrossRef] [PubMed]
- Comber, J.D.; Philip, R. MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines 2014, 2, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2016, 45, D158–D169. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD – Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kouranov, A.; Xie, L.; de la Cruz, J.; Chen, L.; Westbrook, J.; Bourne, P.E.; Berman, H.M. The RCSB PDB information portal for structural genomics. Nucleic Acids Res. 2006, 34, D302–D305. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2020, 28, 235–242. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific glycan analysis of the SARS-CoV-2 spike. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Watanabe, Y.; Berndsen, Z.T.; Raghwani, J.; Seabright, G.E.; Allen, J.D.; Pybus, O.G.; McLellan, J.S.; Wilson, I.A.; Bowden, T.A.; Ward, A.B.; et al. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat. Commun. 2020, 11, 26883. [Google Scholar] [CrossRef]
- Yang, T.J.; Chang, Y.C.; Ko, T.P.; Draczkowski, P.; Chien, Y.C.; Chang, Y.C.; Wu, K.P.; Khoo, K.H.; Chang, H.W.; Hsu, S.T.D. Cryo-EM analysis of a feline coronavirus spike protein reveals a unique structure and camouflaging glycans. Proc. Natl. Acad. Sci. USA 2020, 117, 1438–1446. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Xiong, X.; Park, Y.J.; Tortorici, M.A.; Snijder, J.; Quispe, J.; Cameroni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected Receptor Functional Mimicry Elucidates Activation of Coronavirus Fusion. Cell 2019, 176, 1026–1039. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat. Commun. 2020, 11, 3618. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B Cell Epitopes | T Cell Epitopes | ||||
|---|---|---|---|---|---|
| Protein Name | #Unique Sequences | Conserved | Candidate | Conserved | Candidate |
| Spike Glycoprotein | 4595 | 50 | 152 | 114 | 29 |
| Nucleoprotein | 1737 | 52 | 62 | 65 | 54 |
| Membrane Protein | 285 | 8 | 16 | 18 | 9 |
| Protein 3a | 4 | 1 | 1 | 21 | 13 |
| Envelope Protein | 1 | 1 | 0 | 3 | 0 |
| ORF6 Protein | 109 | 0 | 0 | 5 | 1 |
| Protein 7a | 217 | 0 | 1 | 9 | 3 |
| Protein 7b | 98 | 0 | 0 | 2 | 1 |
| Protein 9b | 7 | 0 | 1 | 0 | 0 |
| Replicase Polyprotein 1a * | 11,509 | 0 | 4 | 3 | 0 |
| Replicase Polyprotein 1ab * | 795 | 0 | 0 | 39 | 0 |
| EPITOPE | NUMBER GENOMES | HOMOLOGOUS EPITOPES and CLUSTER CHANGE RATE | START POS | PARENT EPITOPES |
|---|---|---|---|---|
| Nucleo B cell Epitopes | ||||
| LLPAAD | 61,007 | 0.0 | 393 | N.A |
| QGTDYKHW | 61,004 | [] 0.0 | 293 | N.A |
| SKKPRQKRTATKAYNV | 60,996 | [SKKPRQKRTATKQYNV] 0.5 | 254 | SKKPRQKRTATKQYNV |
| KRTATKAYNVTQAFGRR | 60,981 | [KRTATKQYNVTQAFGRR] 0.5 | 260 | KRTATKQYNVTQAFGRR |
| TKAYNVTQAFGRRGP | 60,980 | [TKQYNVTQAFGRRG] 0.5 | 264 | TKQYNVTQAFGRRGP |
| QFAPSASAFFGMSRIGM | 60,978 | [] 0.0 | 305 | N.A |
| KHWPQIAQFAPSASAFF | 60,950 | [] 0.0 | 298 | N.A |
| KKSAAEASKKPRQKRTA | 60,947 | [] 0.0 | 247 | N.A |
| YNVTQAFGRRGPEQTQGNF | 60,941 | [VTQAFGRRGPEQTQGNFGDQ] 0.0 | 267 | N.A |
| VTQAFGRRGPEQTQGNFGDQ | 60,920 | [YNVTQAFGRRGPEQTQGNF] 0.0 | 269 | N.A |
| Nucleo T cell Epitopes | ||||
| ILLNKHID * | 61,092 | [ILLNKHIDA] 0.0 | 350 | N.A |
| ILLNKHIDA * | 61,069 | [ILLNKHID] 0.0 | 350 | N.A |
| LALLLLDRL * | 61,063 | [LLLDRLNQL, GDAALALLLLDRLNQL] 0.0 | 218 | N.A |
| FSKQLQQSM * | 61,063 | [] 0.0 | 402 | N.A |
| GMSRIGMEV * | 61,025 | [] 0.0 | 315 | N.A |
| KAYNVTQAF * | 61,024 | [ TKQYNVTQAF] 0.0 | 265 | N.A |
| LLLDRLNQL * | 61,012 | [LALLLLDRL, GDAALALLLLDRLNQL] 0.0 | 221 | N.A |
| AQFAPSASAFFGMSR | 60,996 | [ AQFAPSASAFFGMSRIGM] 0.0 | 304 | N.A |
| SKKPRQKRTATKAYNV | 60,996 | [SKKPRQKRTATKQYNV] 0.5 | 254 | SKKPRQKRTATKQYNV |
| QKRTATKAYNVTQAF | 60,988 | [QKRTATKQYNVTQAF, RQKRTATKAYNVIQAFGRRG] 0.66 | 259 | QKRTATKQYNVTQAF |
| EPITOPE | NUMBER GENOMES | HOMOLOGOUS EPITOPES and CLUSTER CHANGE RATE | START POS | PARENT EPITOPES |
|---|---|---|---|---|
| Spike B cell Epitopes | ||||
| ILSRLDKVEAEVQIDRL | 61,222 | [ILSRLDKVEAEVQIDRL] 0.0 | 979 | N.A |
| DFCGKGYHLMSFPQSAP | 61,215 | [DFCGKGYHLMSFPQSAP] 1.0 | 1040 | DFCGKGYHLMSFPQAAP |
| MAYRFNGIGVTQNVLYE | 61,213 | [MAYRFNGIGVTQNVLYE] 0.0 | 901 | N.A |
| VLGQSKRVDFCGKGYHL | 61,212 | [VLGQSKRVDFCGKGYHL] 0.0 | 1032 | N.A |
| AISSVLNDILSRLDKVE | 61,211 | [AISSVLNDILSRLDKVE] 0.0 | 971 | N.A |
| GSFCTQLN | 61,209 | [GSFCTQLN] 0.0 | 756 | N.A |
| EAEVQIDRLITGRLQSL | 61,205 | [EAEVQIDRLITGRLQSL] 0.0 | 987 | N.A |
| KQLSSNFGAISSVLNDI | 61,205 | [KQLSSNFGAISSVLNDI] 0.0 | 963 | N.A |
| SLQTYVTQQLIRAAEIR | 61,200 | [SLQTYVTQQLIRAAEIR] 0.0 | 1002 | N.A |
| LMSFPQSAPHGVVFLHV | 61,199 | [LMSFPQSAPHGVVFLHV] 1.0 | 1048 | LMSFPQAAPHGVVFLHV |
| Spike T cell Epitopes | ||||
| FPQSAPHGVVF * | 61,227 | [] 0.0 | 1051 | N.A |
| RVDFCGKGY * | 61,225 | [] 0.0 | 1038 | N.A |
| VLNDILSRL * | 61,225 | [ SVLNDILSRL] 0.0 | 975 | N.A |
| SVLNDILSRL * | 61,224 | [VLNDILSRL] 0.0 | 974 | N.A |
| YHLMSFPQSA * | 61,223 | [] 0.0 | 1046 | N.A |
| MAYRFNGIGVTQNVLY | 61,216 | [] 0.0 | 901 | N.A |
| ALNTLVKQL * | 61,212 | [AQALNTLVKQL] 0.0 | 957 | N.A |
| AQALNTLVKQL * | 61,211 | [ALNTLVKQL] 0.0 | 955 | N.A |
| SFPQSAPHGVVFLHV | 61,210 | [LMSFPQSAPHGVVFLHV] 0.5 | 1050 | N.A |
| LITGRLQSL * | 61,209 | [] 0.0 | 995 | N.A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Agarwal, A.; Beck, K.L.; Capponi, S.; Kunitomi, M.; Nayar, G.; Seabolt, E.; Mahadeshwar, G.; Bianco, S.; Mukherjee, V.; Kaufman, J.H. Predicting Epitope Candidates for SARS-CoV-2. Viruses 2022, 14, 1837. https://doi.org/10.3390/v14081837
Agarwal A, Beck KL, Capponi S, Kunitomi M, Nayar G, Seabolt E, Mahadeshwar G, Bianco S, Mukherjee V, Kaufman JH. Predicting Epitope Candidates for SARS-CoV-2. Viruses. 2022; 14(8):1837. https://doi.org/10.3390/v14081837
Chicago/Turabian StyleAgarwal, Akshay, Kristen L. Beck, Sara Capponi, Mark Kunitomi, Gowri Nayar, Edward Seabolt, Gandhar Mahadeshwar, Simone Bianco, Vandana Mukherjee, and James H. Kaufman. 2022. "Predicting Epitope Candidates for SARS-CoV-2" Viruses 14, no. 8: 1837. https://doi.org/10.3390/v14081837