
Pharmacophore-Model-Based Drug Repurposing for the Identification of the Potential Inhibitors Targeting the Allosteric Site in Dengue Virus NS5 RNA-Dependent RNA Polymerase

 , , ,
, , ,  and
and 
Abstract
:
1. Introduction
2. Methodology
2.1. Structure Collection and Preparation
2.2. Pharmacophore Modelling and Ligand-Based Screening
2.3. Structure-Based Virtual Screening
2.4. MM/GBSA Binding Free Energy
2.5. Molecular Dynamics Simulation
2.6. End-Point Binding Free Energy Calculation
3. Results
3.1. Pharmacophore Model Generation and Screening
3.2. Structure-Based Screening
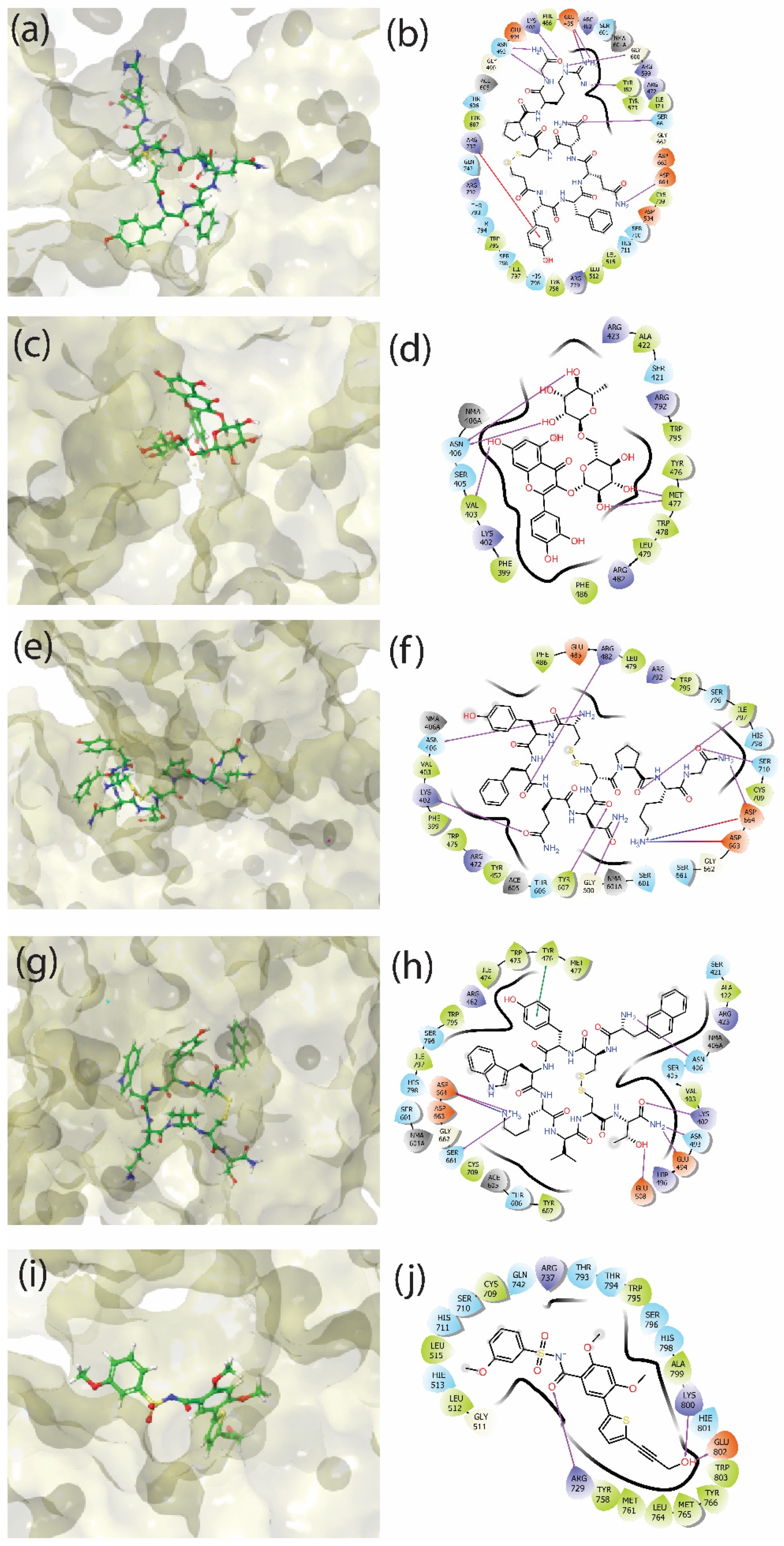
3.3. Molecular Interactions
3.4. Explicit Molecular Dynamics Simulations
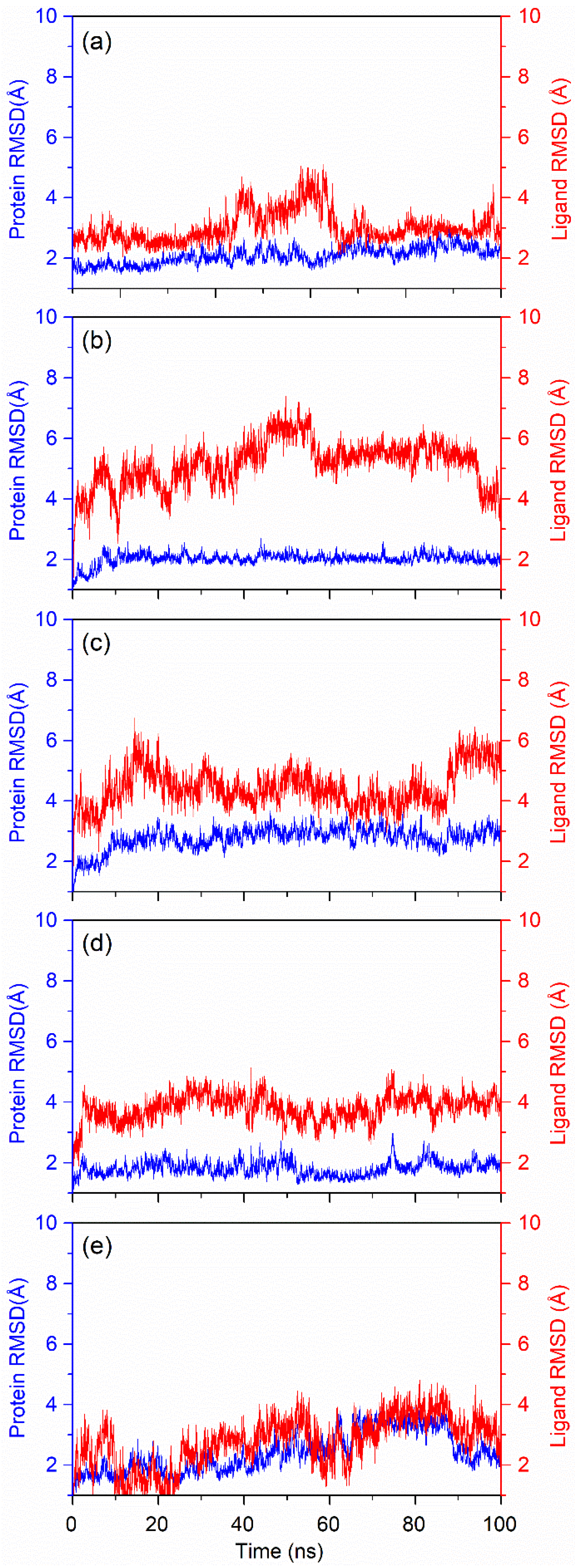
3.4.1. RMSD Fluctuation
3.4.2. RMSF Fluctuation
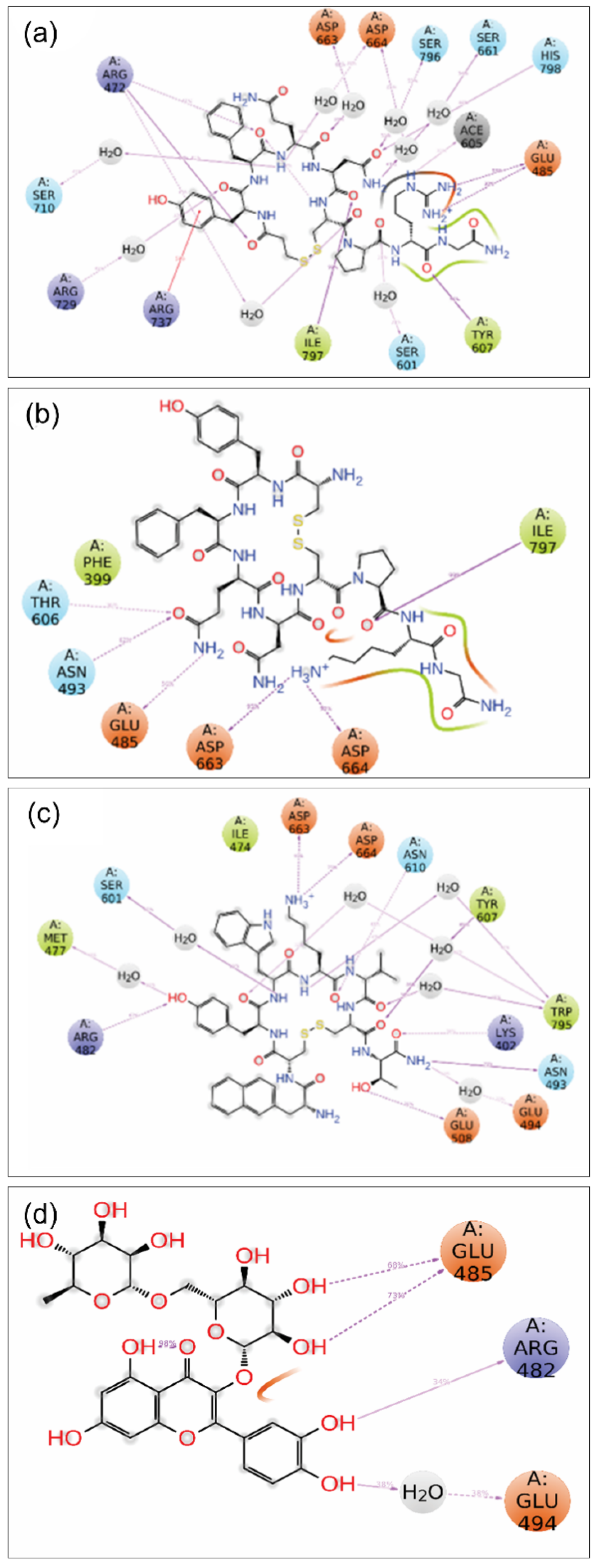
3.4.3. MD Trajectory Interaction
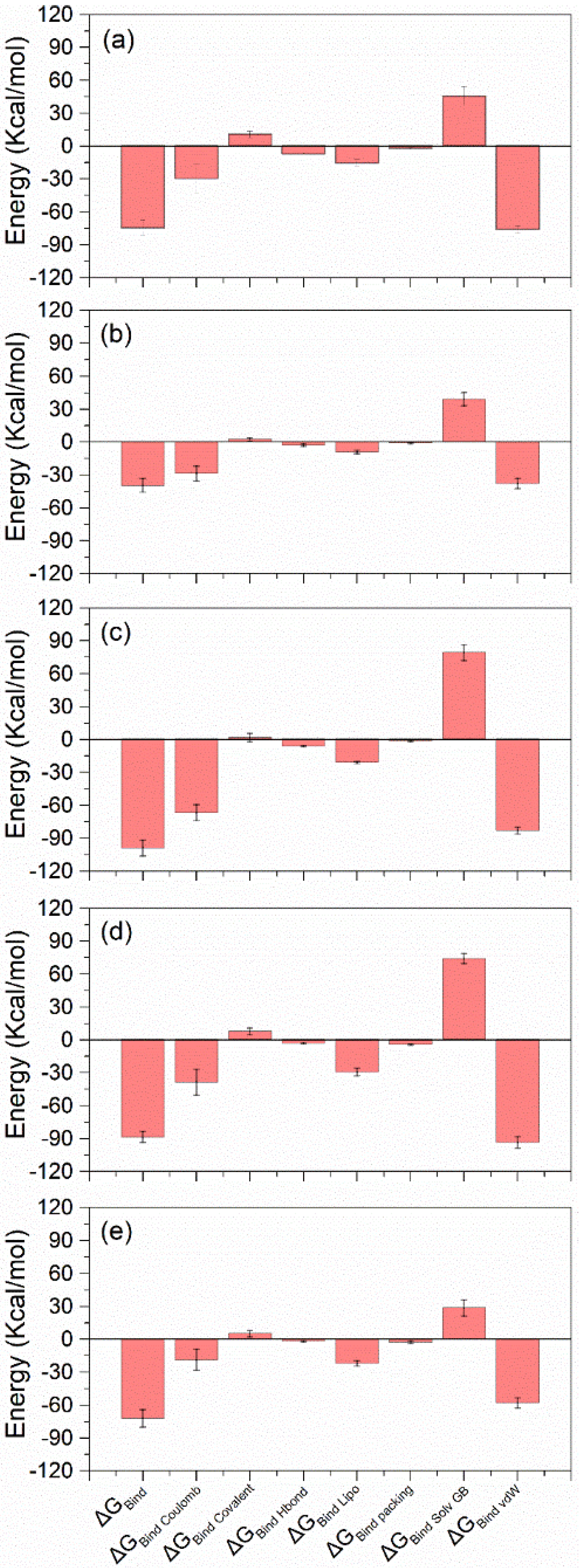
3.4.4. MD Trajectory MM/GBSA
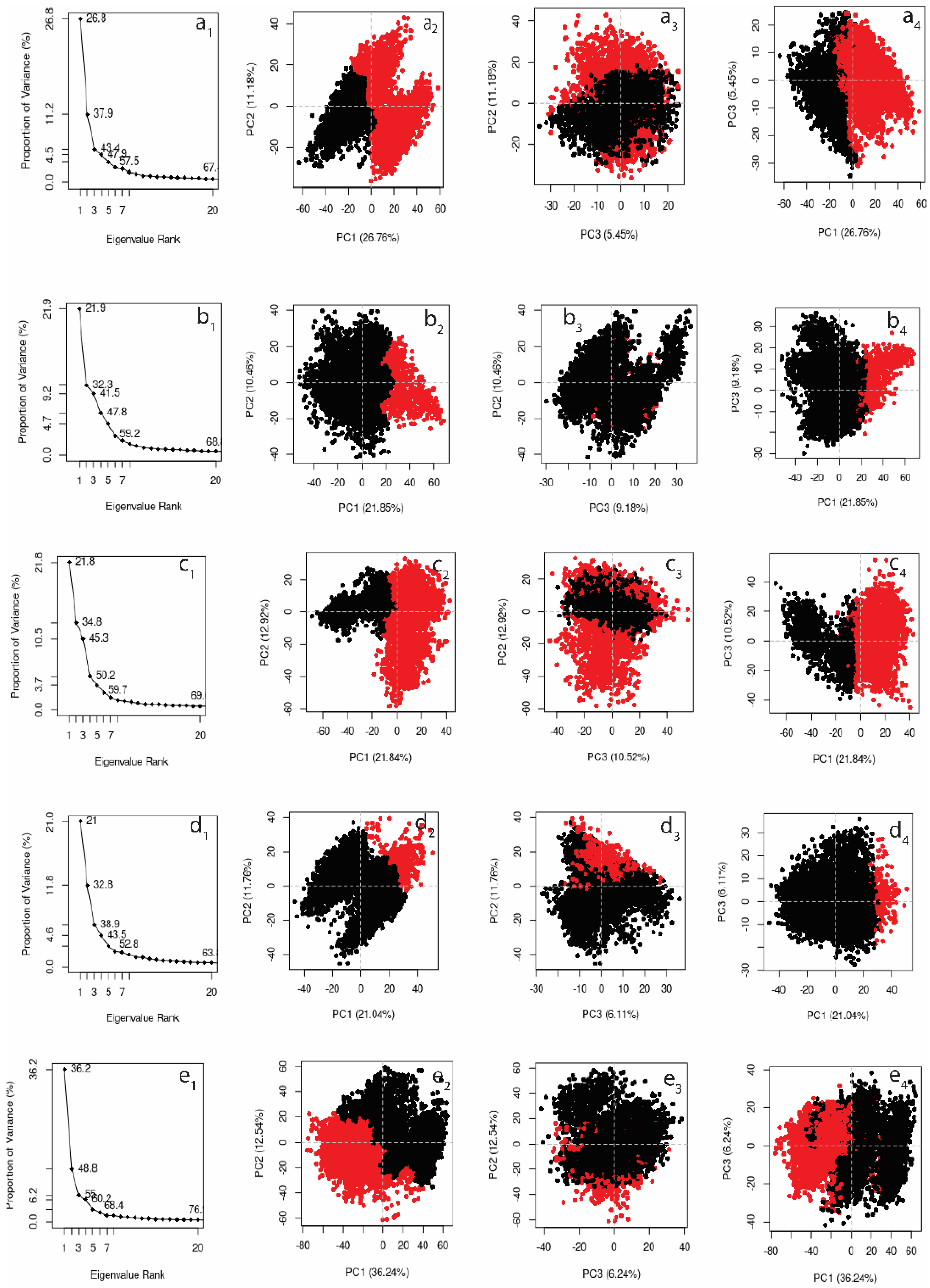
3.4.5. Principal Component Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhatt, S.; Gething, P.W.; Brady, O.J.; Messina, J.P.; Farlow, A.W.; Moyes, C.L.; Drake, J.M.; Brownstein, J.S.; Hoen, A.G.; Sankoh, O.; et al. The global distribution and burden of dengue. Nature 2013, 496, 504–507. [Google Scholar] [CrossRef]
- Capeding, M.R.; Tran, N.H.; Hadinegoro, S.R.S.; Ismail, H.I.H.J.M.; Chotpitayasunondh, T.; Chua, M.N.; Luong, C.Q.; Rusmil, K.; Wirawan, D.N.; Nallusamy, R.; et al. Clinical efficacy and safety of a novel tetravalent dengue vaccine in healthy children in Asia: A phase 3, randomised, observer-masked, placebo-controlled trial. Lancet 2014, 384, 1358–1365. [Google Scholar] [CrossRef]
- Sabchareon, A.; Wallace, D.; Sirivichayakul, C.; Limkittikul, K.; Chanthavanich, P.; Suvannadabba, S.; Jiwariyavej, V.; Dulyachai, W.; Pengsaa, K.; Wartel, T.A.; et al. Protective efficacy of the recombinant, live-attenuated, CYD tetravalent dengue vaccine in Thai schoolchildren: A randomised, controlled phase 2b trial. Lancet 2012, 380, 1559–1567. [Google Scholar] [CrossRef]
- Lim, S.P.; Wang, Q.-Y.; Noble, C.G.; Chen, Y.-L.; Dong, H.; Zou, B.; Yokokawa, F.; Nilar, S.; Smith, P.; Beer, D.; et al. Ten years of dengue drug discovery: Progress and prospects. Antivir. Res. 2013, 100, 500–519. [Google Scholar] [CrossRef]
- Boldescu, V.; Behnam, M.A.M.; Vasilakis, N.; Klein, C.D. Broad-spectrum agents for flaviviral infections: Dengue, zika and beyond. Nat. Rev. Drug Discov. 2017, 16, 565–586. [Google Scholar] [CrossRef] [Green Version]
- Low, J.G.; Gatsinga, R.; Vasudevan, S.G.; Sampath, A. Dengue antiviral development: A continuing journey. In Dengue and Zika: Control and Antiviral Treatment Strategies; Hilgenfeld, R., Vasudevan, S.G., Eds.; Advances in Experimental Medicine and Biology; Springer: Singapore, 2018; Volume 1062, pp. 319–332. ISBN 978-981-10-8726-4. [Google Scholar]
- Troost, B.; Smit, J.M. Recent advances in antiviral drug development towards dengue virus. Curr. Opin. Virol. 2020, 43, 9–21. [Google Scholar] [CrossRef]
- Mackenzie, J. Wrapping things up about virus RNA replication. Traffic 2005, 6, 967–977. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kääriäinen, L. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 2005, 285, 139–173. [Google Scholar] [CrossRef]
- Ackermann, M.; Padmanabhan, R. De novo synthesis of RNA by the dengue virus RNA-dependent RNA polymerase exhibits temperature dependence at the initiation but not elongation phase. J. Biol. Chem. 2001, 276, 39926–39937. [Google Scholar] [CrossRef] [Green Version]
- Maddipati, V.C.; Mittal, L.; Mantipally, M.; Asthana, S.; Bhattacharyya, S.; Gundla, R. A review on the progress and prospects of dengue drug discovery targeting NS5 RNA- dependent RNA polymerase. Curr. Pharm. Des. 2020, 26, 4386–4409. [Google Scholar] [CrossRef]
- Zhao, Y.; Soh, T.S.; Zheng, J.; Chan, K.W.K.; Phoo, W.W.; Lee, C.C.; Tay, M.Y.F.; Swaminathan, K.; Cornvik, T.C.; Lim, S.P.; et al. A crystal structure of the dengue virus NS5 protein reveals a novel inter-domain interface essential for protein flexibility and virus replication. PLoS Pathog. 2015, 11, e1004682. [Google Scholar] [CrossRef]
- Jin, Z.; Deval, J.; Johnson, K.A.; Swinney, D.C. Characterization of the elongation complex of dengue virus RNA polymerase: Assembly, kinetics of nucleotide incorporation, and fidelity. J. Biol. Chem. 2011, 286, 2067–2077. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.P.; Noble, C.G.; Seh, C.C.; Soh, T.S.; El Sahili, A.; Chan, G.K.Y.; Lescar, J.; Arora, R.; Benson, T.; Nilar, S.; et al. Potent allosteric dengue virus NS5 polymerase inhibitors: Mechanism of action and resistance profiling. PLoS Pathog. 2016, 12, e1005737. [Google Scholar] [CrossRef] [Green Version]
- Yokokawa, F.; Nilar, S.; Noble, C.G.; Lim, S.P.; Rao, R.; Tania, S.; Wang, G.; Lee, G.; Hunziker, J.; Karuna, R.; et al. Discovery of potent non-nucleoside inhibitors of dengue viral RNA-dependent RNA polymerase from a fragment hit using structure-based drug design. J. Med. Chem. 2016, 59, 3935–3952. [Google Scholar] [CrossRef]
- Noble, C.G.; Lim, S.P.; Arora, R.; Yokokawa, F.; Nilar, S.; Seh, C.C.; Wright, S.K.; Benson, T.E.; Smith, P.W.; Shi, P.-Y. A conserved pocket in the dengue virus polymerase identified through fragment-based screening. J. Biol. Chem. 2016, 291, 8541–8548. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Espiritu, M.V.; Abraham, J.; Thorpe, I.F. Computational predictions suggest that structural similarity in viral polymerases may lead to comparable allosteric binding sites. Virus Res. 2016, 222, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Van Tongeren, S.A.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G.; et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014, 508, 402–405. [Google Scholar] [CrossRef] [Green Version]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef]
- Crotty, S.; Maag, D.; Arnold, J.J.; Zhong, W.; Lau, J.Y.N.; Hong, Z.; Andino, R.; Cameron, C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 2000, 6, 1375–1379. [Google Scholar] [CrossRef]
- Tian, L.; Qiang, T.; Liang, C.; Ren, X.; Jia, M.; Zhang, J.; Li, J.; Wan, M.; YuWen, X.; Li, H.; et al. RNA-dependent RNA polymerase (RdRp) inhibitors: The current landscape and repurposing for the COVID-19 pandemic. Eur. J. Med. Chem. 2021, 213, 113201. [Google Scholar] [CrossRef]
- Sofia, M.J.; Chang, W.; Furman, P.A.; Mosley, R.T.; Ross, B.S. Nucleoside, Nucleotide, and Non-Nucleoside Inhibitors of Hepatitis C Virus NS5B RNA-Dependent RNA-Polymerase. J. Med. Chem. 2012, 55, 2481–2531. [Google Scholar] [CrossRef]
- Chen, H.; Zhang, Z.; Zhang, J. In silico drug repositioning based on integrated drug targets and canonical correlation analysis. BMC Med. Genom. 2022, 15, 48. [Google Scholar] [CrossRef]
- Botta, L.; Rivara, M.; Zuliani, V.; Radi, M. Drug repurposing approaches to fight dengue virus infection and related diseases. Front. Biosci. Landmark Ed. 2018, 23, 997–1019. [Google Scholar] [CrossRef] [Green Version]
- Low, J.G.H.; Ooi, E.E.; Vasudevan, S.G. Current status of dengue therapeutics research and development. J. Infect. Dis. 2017, 215, S96–S102. [Google Scholar] [CrossRef] [Green Version]
- Cheung, Y.Y.; Chen, K.C.; Chen, H.; Seng, E.K.; Chu, J.J.H. Antiviral activity of lanatoside C against dengue virus infection. Antivir. Res. 2014, 111, 93–99. [Google Scholar] [CrossRef]
- Montes-Grajales, D.; Puerta-Guardo, H.; Espinosa, D.A.; Harris, E.; Caicedo-Torres, W.; Olivero-Verbel, J.; Martínez-Romero, E. In silico drug repurposing for the identification of potential candidate molecules against arboviruses infection. Antivir. Res. 2020, 173, 104668. [Google Scholar] [CrossRef]
- Amemiya, T.; Horimoto, K.; Fukui, K. Application of multiple omics and network projection analyses to drug repositioning for pathogenic mosquito-borne viruses. Sci. Rep. 2021, 11, 10136. [Google Scholar] [CrossRef]
- Pathak, N.; Lai, M.-L.; Chen, W.-Y.; Hsieh, B.-W.; Yu, G.-Y.; Yang, J.-M. Pharmacophore anchor models of flaviviral NS3 proteases lead to drug repurposing for DENV infection. BMC Bioinform. 2017, 18, 548. [Google Scholar] [CrossRef] [Green Version]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2018-3: Protein Preparation Wizard; Epik, Schrödinger, LLC.: New York, NY, USA, 2018.
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for PK a prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, V.D.; Arya, A.; Yadav, P.; Kumar, R.; Kumar, V.; Raghava, G.P.S. DenvInD: Dengue virus inhibitors database for clinical and molecular research. Brief. Bioinform. 2021, 22, bbaa098. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-3: LigPrep; Schrödinger, LLC.: New York, NY, USA, 2018.
- Huang, R.; Southall, N.; Wang, Y.; Yasgar, A.; Shinn, P.; Jadhav, A.; Nguyen, D.-T.; Austin, C.P. The NCGC pharmaceutical collection: A comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci. Transl. Med. 2011, 3, 80ps16. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.L.; Smondyrev, A.M.; Rao, S.N. PHASE: A novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 2006, 67, 370–372. [Google Scholar] [CrossRef]
- Dixon, S.L.; Smondyrev, A.M.; Knoll, E.H.; Rao, S.N.; Shaw, D.E.; Friesner, R.A. PHASE: A new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. methodology and preliminary results. J. Comput. Aided Mol. Des. 2006, 20, 647–671. [Google Scholar] [CrossRef]
- Schrödinger Release 2021-4: Phase; Schrödinger, LLC.: New York, NY, USA, 2021.
- Friesner, R.A.; Murphy, R.B.; Repasky, M.P.; Frye, L.L.; Greenwood, J.R.; Halgren, T.A.; Sanschagrin, P.C.; Mainz, D.T. Extra precision glide: Docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J. Med. Chem. 2006, 49, 6177–6196. [Google Scholar] [CrossRef] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Schrödinger Release 2018-3: Glide; Schrödinger, LLC.: New York, NY, USA, 2018.
- Schrödinger Release 2018-3: Prime; Schrödinger, LLC.: New York, NY, USA, 2018.
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef] [Green Version]
- Bowers, K.J.; Chow, D.E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D.; et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE Conference on Supercomputing (SC’06), Tampa, FL, USA, 11–17 November 2006; IEEE: Manhattan, NY, USA, 2006; p. 43. [Google Scholar]
- Schrödinger Release 2020-4: Desmond Molecular Dynamics System; Maestro-Desmond Interoperability Tools; D.E. Shaw Research: New York, NY, USA; Schrödinger: New York, NY, USA, 2020.
- Kotnik, P.; Nielsen, J.; Kwon, T.-H.; Krzisnik, C.; Frøkiaer, J.; Nielsen, S. Altered expression of COX-1, COX-2, and MPGES in rats with nephrogenic and central diabetes insipidus. Am. J. Physiol. Renal Physiol. 2005, 288, F1053–F1068. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-P.; Wu, P.-P.; Hou, Y.-C.; Lin, S.-P.; Tsai, S.-Y.; Chen, C.-T.; Chao, P.-D.L. Quercetin and rutin reduced the bioavailability of cyclosporine from neoral, an immunosuppressant, through activating P-glycoprotein and CYP 3A4. J. Agric. Food Chem. 2011, 59, 4644–4648. [Google Scholar] [CrossRef] [PubMed]
- Madon, J.; Hagenbuch, B.; Landmann, L.; Meier, P.J.; Stieger, B. Transport function and hepatocellular localization of Mrp6 in rat liver. Mol. Pharmacol. 2000, 57, 634–641. [Google Scholar] [CrossRef]
- Trocóniz, I.F.; Cendrós, J.-M.; Peraire, C.; Ramis, J.; Garrido, M.J.; Boscani, P.F.; Obach, R. Population pharmacokinetic analysis of lanreotide autogel in healthy subjects: Evidence for injection interval of up to 2 months. Clin. Pharmacokinet. 2009, 48, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Bourjot, M.; Leyssen, P.; Eydoux, C.; Guillemot, J.C.; Canard, B.; Rasoanaivo, P.; Guéritte, F.; Litaudon, M. Flacourtosides A-F, phenolic glycosides isolated from Flacourtia ramontchi. J. Nat. Prod. 2012, 75, 752–758. [Google Scholar] [CrossRef]
- Vincetti, P.; Caporuscio, F.; Kaptein, S.; Gioiello, A.; Mancino, V.; Suzuki, Y.; Yamamoto, N.; Crespan, E.; Lossani, A.; Maga, G.; et al. Discovery of Multitarget Antivirals Acting on Both the Dengue Virus NS5-NS3 Interaction and the Host Src/Fyn Kinases. J. Med. Chem. 2015, 58, 4964–4975. [Google Scholar] [CrossRef]
- Allard, P.M.; Dau, E.T.; Eydoux, C.; Guillemot, J.C.; Dumontet, V.; Poullain, C.; Canard, B.; Guéritte, F.; Litaudon, M. Alkylated flavanones from the bark of Cryptocarya chartacea as dengue virus NS5 polymerase inhibitors. J. Nat. Prod. 2011, 74, 2446–2453. [Google Scholar] [CrossRef]
- Bhakat, S.; Karubiu, W.; Jayaprakash, V.; Soliman, M.E. A perspective on targeting non-structural proteins to combat neglected tropical diseases: Dengue, West Nile and Chikungunya viruses. Eur. J. Med. Chem. 2014, 87, 677–702. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S. No. | PubChem ID | HBA | HBD | Molecular Mass | Number of Rings | Aromatic Bonds |
|---|---|---|---|---|---|---|
| 1 | 6439576 | 60 | 3 | 618.842 | 6 | 6 |
| 2 | 21672233 | 28 | 4 | 376.357 | 3 | 12 |
| 3 | 44577154 | 37 | 5 | 526.489 | 4 | 18 |
| 4 | 46898022 | 26 | 3 | 375.406 | 4 | 16 |
| 5 | 49799036 | 32 | 4 | 418.473 | 4 | 16 |
| 6 | 49799133 | 28 | 3 | 339.395 | 4 | 16 |
| 7 | 56834067 | 32 | 3 | 458.502 | 4 | 18 |
| 8 | 56834069 | 48 | 4 | 660.751 | 5 | 24 |
| 9 | 56834070 | 48 | 4 | 660.751 | 5 | 24 |
| 10 | 56834169 | 48 | 4 | 660.751 | 5 | 24 |
| 11 | 56834170 | 48 | 4 | 660.751 | 5 | 24 |
| 12 | 56834171 | 32 | 3 | 458.502 | 4 | 18 |
| 13 | 56834172 | 32 | 3 | 458.502 | 4 | 18 |
| 14 | 56834173 | 32 | 3 | 458.502 | 4 | 18 |
| 15 | 56834283 | 32 | 3 | 458.502 | 4 | 18 |
| 16 | 57409245 | 49 | 6 | 670.614 | 5 | 18 |
| 17 | 57409246 | 50 | 7 | 698.624 | 5 | 18 |
| 18 | 57409247 | 47 | 6 | 680.609 | 5 | 18 |
| 19 | 60165190 | 43 | 5 | 646.594 | 5 | 24 |
| 20 | 70683874 | 47 | 6 | 680.609 | 5 | 18 |
| 21 | 118717693 | 26 | 5 | 538.458 | 6 | 34 |
| 22 | 118779901 | 22 | 1 | 301.301 | 4 | 11 |
| 23 | 118797900 | 16 | 2 | 276.308 | 2 | 11 |
| 24 | 118797902 | 23 | 2 | 379.451 | 2 | 11 |
| 25 | 121232415 | 29 | 2 | 487.545 | 3 | 17 |
| 26 | 127043014 | 16 | 2 | 288.318 | 2 | 11 |
| 27 | 127043015 | 18 | 2 | 302.345 | 2 | 11 |
| 28 | 127043016 | 17 | 2 | 312.346 | 3 | 16 |
| 29 | 127043018 | 19 | 2 | 324.397 | 3 | 16 |
| 30 | 127043019 | 23 | 2 | 379.451 | 2 | 11 |
| 31 | 127043024 | 25 | 2 | 441.52 | 3 | 17 |
| 32 | 127043025 | 26 | 2 | 457.519 | 3 | 17 |
| 33 | 127043211 | 25 | 2 | 491.964 | 3 | 17 |
| 34 | 127043212 | 27 | 2 | 492.567 | 4 | 22 |
| 35 | 127043361 | 15 | 2 | 310.753 | 2 | 11 |
| 36 | 127044830 | 20 | 4 | 410.804 | 4 | 23 |
| 37 | 127044864 | 18 | 2 | 302.345 | 2 | 11 |
| 38 | 127045349 | 15 | 2 | 355.204 | 2 | 11 |
| 39 | 137243533 | 18 | 2 | 342.369 | 3 | 16 |
| 40 | 57409350 | 52 | 5 | 784.715 | 6 | 24 |
| 41 | 135434165 | 32 | 7 | 507.181 | 3 | 10 |
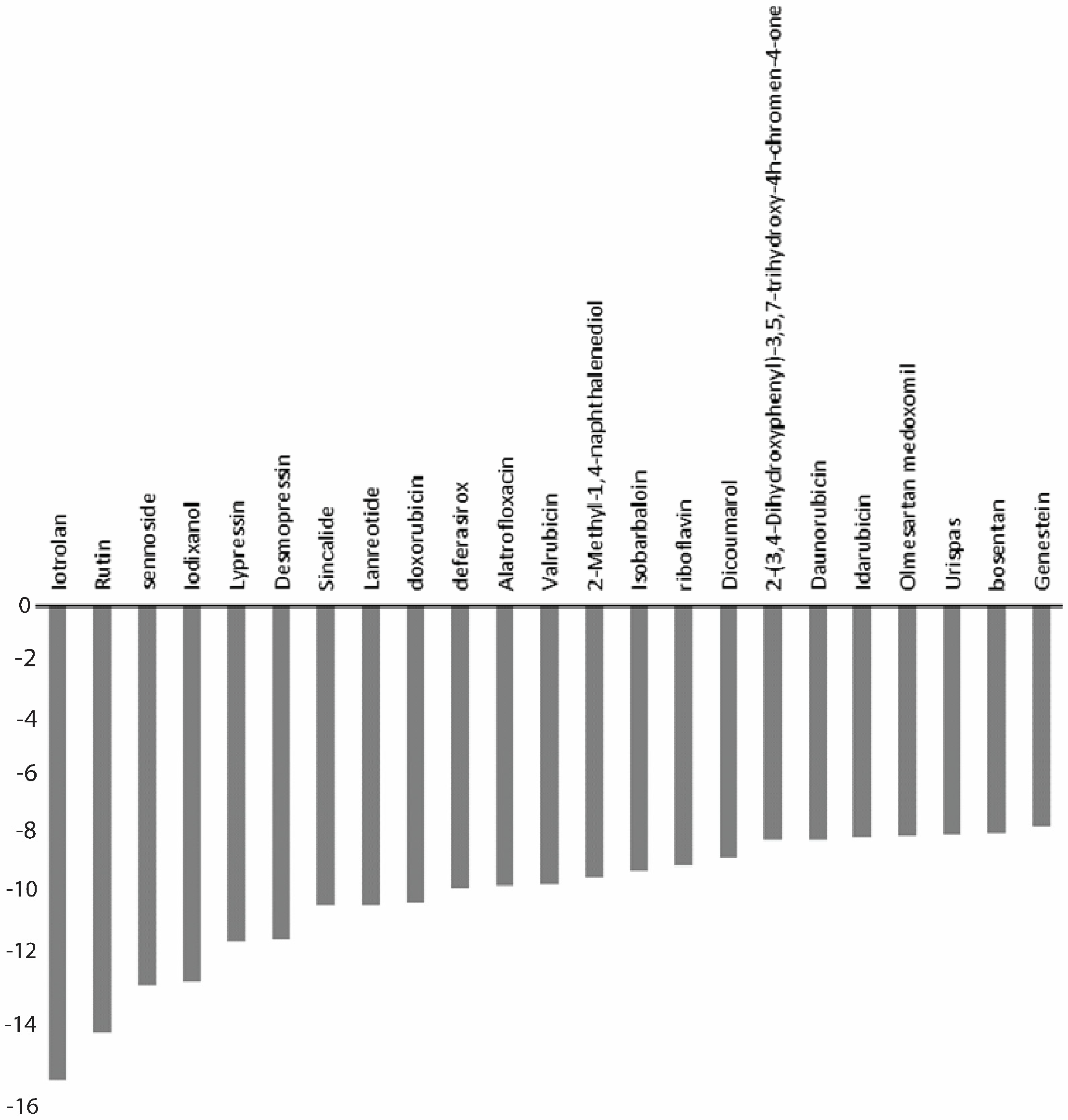
| S.No. | Drugs | Docking Score (kcal/mol) | XP GScore (kcal/mol) | MMGBSA ΔGBind (kcal/mol) |
|---|---|---|---|---|
| 1 | Iotrolan | −14.071 | −14.965 | −88.58 |
| 2 | Desmopressin | −10.527 | −10.527 | −69.77 |
| 3 | Rutin | −13.435 | −13.463 | −67.06 |
| 4 | Lypressin | −9.84 | −10.597 | −67.65 |
| 5 | Lanreotide | −8.727 | −9.436 | −64.7 |
| 6 | Bosentan | −7.194 | −7.194 | −64.61 |
| 7 | Sennoside | −11.979 | −11.987 | −62.2 |
| 8 | Valrubicin | −8.814 | −8.814 | −58.41 |
| 9 | Sincalide | −9.432 | −9.449 | −53.1 |
| 10 | Riboflavin | −8.185 | −8.185 | −48.71 |
| 11 | Daunorubicin | −7.349 | −7.376 | −47.72 |
| 12 | Idarubicin | −7.251 | −7.3 | −47.51 |
| 13 | Deferasirox | −8.929 | −8.935 | −46.56 |
| 14 | 2-(3,4-Dihydroxyphenyl)-3,5,7-Trihydroxy-4h-chromen-4-one | −7.347 | −7.379 | −45.74 |
| 15 | Genestein | −6.912 | −6.937 | −43 |
| 16 | Olmesartan medoxomil | −7.242 | −7.283 | −42.33 |
| 17 | Isobarbaloin | −8.356 | −8.356 | −39.58 |
| 18 | Doxorubicin | −9.315 | −9.342 | −38.01 |
| 19 | Urispas | −7.21 | −7.21 | −36.82 |
| 20 | Alatrofloxacin | −7.455 | −8.841 | −30.63 |
| 21 | Iodixanol | −11.839 | −11.839 | −29.7 |
| 22 | Dicoumarol | −7.82 | −7.935 | −11.43 |
| 23 | 2-Methyl-1,4-naphthalenediol | −7.121 | −8.539 | −6.99 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, S.; Bajrai, L.H.; Faizo, A.A.; Khateb, A.M.; Alkhaldy, A.A.; Rana, R.; Azhar, E.I.; Dwivedi, V.D. Pharmacophore-Model-Based Drug Repurposing for the Identification of the Potential Inhibitors Targeting the Allosteric Site in Dengue Virus NS5 RNA-Dependent RNA Polymerase. Viruses 2022, 14, 1827. https://doi.org/10.3390/v14081827
Kumar S, Bajrai LH, Faizo AA, Khateb AM, Alkhaldy AA, Rana R, Azhar EI, Dwivedi VD. Pharmacophore-Model-Based Drug Repurposing for the Identification of the Potential Inhibitors Targeting the Allosteric Site in Dengue Virus NS5 RNA-Dependent RNA Polymerase. Viruses. 2022; 14(8):1827. https://doi.org/10.3390/v14081827
Chicago/Turabian StyleKumar, Sanjay, Leena H. Bajrai, Arwa A. Faizo, Aiah M. Khateb, Areej A. Alkhaldy, Rashmi Rana, Esam I. Azhar, and Vivek Dhar Dwivedi. 2022. "Pharmacophore-Model-Based Drug Repurposing for the Identification of the Potential Inhibitors Targeting the Allosteric Site in Dengue Virus NS5 RNA-Dependent RNA Polymerase" Viruses 14, no. 8: 1827. https://doi.org/10.3390/v14081827