
Beyond Amyloid Fibers: Accumulation, Biological Relevance, and Regulation of Higher-Order Prion Architectures


Abstract
:1. Introduction
2. The Functional Properties of Amyloids Assembled In Vitro or In Vivo Are Similar but Distinct
3. Prion and Prion-like Proteins Access a Range of Aggregate States with the Potential for Biological Impact
4. De Novo Induction of [PSI+] Is Associated with Higher-Order Sup35 Architecture of Unknown Origin and Function
5. Assembly State Impacts Stability of the Prion State
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and Chemical Approaches to Diseases of Proteostasis Deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular Chaperones in Protein Folding and Proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, R.I. Proteotoxic Stress and Inducible Chaperone Networks in Neurodegenerative Disease and Aging. Genes Dev. 2008, 22, 1427–1438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of Cellular Proteostasis in Aging and Disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.N.; Gracheva, E.O.; Richmond, J.E.; Liebman, S.W. Variant-Specific [PSI] Infection Is Transmitted by Sup35 Polymers within [PSI] Aggregates with Heterogeneous Protein Composition. Mol. Biol. Cell 2008, 19, 2433–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balguerie, A.; Dos Reis, S.; Coulary-Salin, B.; Chaignepain, S.; Sabourin, M.; Schmitter, J.-M.; Saupe, S.J. The Sequences Appended to the Amyloid Core Region of the HET-s Prion Protein Determine Higher-Order Aggregate Organization in Vivo. J. Cell Sci. 2004, 117, 2599–2610. [Google Scholar] [CrossRef] [Green Version]
- Benilova, I.; Reilly, M.; Terry, C.; Wenborn, A.; Schmidt, C.; Marinho, A.T.; Risse, E.; Al-Doujaily, H.; Wiggins De Oliveira, M.; Sandberg, M.K.; et al. Highly Infectious Prions Are Not Directly Neurotoxic. Proc. Natl. Acad. Sci. USA 2020, 117, 23815–23822. [Google Scholar] [CrossRef]
- Kawai-Noma, S.; Pack, C.-G.; Kojidani, T.; Asakawa, H.; Hiraoka, Y.; Kinjo, M.; Haraguchi, T.; Taguchi, H.; Hirata, A. In Vivo Evidence for the Fibrillar Structures of Sup35 Prions in Yeast Cells. J. Cell Biol. 2010, 190, 223–231. [Google Scholar] [CrossRef] [Green Version]
- Kryndushkin, D.S.; Alexandrov, I.M.; Ter-Avanesyan, M.D.; Kushnirov, V.V. Yeast [PSI+] Prion Aggregates Are Formed by Small Sup35 Polymers Fragmented by Hsp104. J. Biol. Chem. 2003, 278, 49636–49643. [Google Scholar] [CrossRef] [Green Version]
- Mathur, V.; Hong, J.Y.; Liebman, S.W. Ssa1 Overexpression and [PIN+] Variants Cure [PSI+] by Dilution of Aggregates. J. Mol. Biol. 2009, 390, 155–167. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.-N.; Zhao, X.; Yim, Y.-I.; Todor, H.; Ellerbrock, R.; Reidy, M.; Eisenberg, E.; Masison, D.C.; Greene, L.E. Hsp104 Overexpression Cures Saccharomyces cerevisiae [PSI+] by Causing Dissolution of the Prion Seeds. Eukaryot. Cell 2014, 13, 635–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saibil, H.R.; Seybert, A.; Habermann, A.; Winkler, J.; Eltsov, M.; Perkovic, M.; Castano-Diez, D.; Scheffer, M.P.; Haselmann, U.; Chlanda, P.; et al. Heritable Yeast Prions Have a Highly Organized Three-Dimensional Architecture with Interfiber Structures. Proc. Natl. Acad. Sci. USA 2012, 109, 14906–14911. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Wu, Y.; Jung, G.; Tutar, Y.; Eisenberg, E.; Greene, L.E.; Masison, D.C. Role for Hsp70 Chaperone in Saccharomyces cerevisiae Prion Seed Replication. Eukaryot. Cell 2005, 4, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyedmers, J.; Treusch, S.; Dong, J.; McCaffery, J.M.; Bevis, B.; Lindquist, S. Prion Induction Involves an Ancient System for the Sequestration of Aggregated Proteins and Heritable Changes in Prion Fragmentation. Proc. Natl. Acad. Sci. USA 2010, 107, 8633–8638. [Google Scholar] [CrossRef] [Green Version]
- Tuite, M.F.; Serio, T.R. The Prion Hypothesis: From Biological Anomaly to Basic Regulatory Mechanism. Nat. Rev. Mol. Cell Biol. 2010, 11, 823–833. [Google Scholar] [CrossRef] [Green Version]
- Sindi, S.S.; Serio, T.R. Prion Dynamics and the Quest for the Genetic Determinant in Protein-Only Inheritance. Curr. Opin. Microbiol. 2009, 12, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.Ø.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the Cross-β Spine of Amyloid-like Fibrils. Nature 2005, 435, 773–778. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.S.; Sawaya, M.R. Structural Studies of Amyloid Proteins at the Molecular Level. Annu. Rev. Biochem. 2017, 86, 69–95. [Google Scholar] [CrossRef] [Green Version]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The Amyloid State and Its Association with Protein Misfolding Diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- Wankhede, N.L.; Kale, M.B.; Upaganlawar, A.B.; Taksande, B.G.; Umekar, M.J.; Behl, T.; Abdellatif, A.A.H.; Bhaskaran, P.M.; Dachani, S.R.; Sehgal, A.; et al. Involvement of Molecular Chaperone in Protein-Misfolding Brain Diseases. Biomed. Pharmacother. 2022, 147, 112647. [Google Scholar] [CrossRef]
- Prusiner, S.B. Novel Proteinaceous Infectious Particles Cause Scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Dong, X. Therapeutic Implications of Prion Diseases. Biosaf. Health 2021, 3, 92–100. [Google Scholar] [CrossRef]
- Levkovich, S.A.; Rencus-Lazar, S.; Gazit, E.; Laor Bar-Yosef, D. Microbial Prions: Dawn of a New Era. Trends Biochem. Sci. 2021, 46, 391–405. [Google Scholar] [CrossRef] [PubMed]
- Marrero-Winkens, C.; Sankaran, C.; Schätzl, H. From Seeds to Fibrils and Back: Fragmentation as an Overlooked Step in the Propagation of Prions and Prion-Like Proteins. Biomolecules 2020, 10, 1305. [Google Scholar] [CrossRef]
- Klaips, C.L.; Hochstrasser, M.L.; Langlois, C.R.; Serio, T.R. Spatial Quality Control Bypasses Cell-Based Limitations on Proteostasis to Promote Prion Curing. eLife 2014, 3, e04288. [Google Scholar] [CrossRef]
- Cox, S. Ψ, A Cytoplasmic Suppressor of Super-Suppressor in Yeast. Hereditary 1965, 20, 505–521. [Google Scholar] [CrossRef] [Green Version]
- Paushkin, S.V.; Kushnirov, V.V.; Smirnov, V.N.; Ter-Avanesyan, M.D. Propagation of the Yeast Prion-like [Psi+] Determinant Is Mediated by Oligomerization of the SUP35-Encoded Polypeptide Chain Release Factor. EMBO J. 1996, 15, 3127–3134. [Google Scholar] [CrossRef]
- Ter-Avanesyan, M.D.; Dagkesamanskaya, A.R.; Kushnirov, V.V.; Smirnov, V.N. The SUP35 Omnipotent Suppressor Gene Is Involved in the Maintenance of the Non-Mendelian Determinant [Psi+] in the Yeast Saccharomyces Cerevisiae. Genetics 1994, 137, 671–676. [Google Scholar] [CrossRef]
- Patino, M.M.; Liu, J.-J.; Glover, J.R.; Lindquist, S. Support for the Prion Hypothesis for Inheritance of a Phenotypic Trait in Yeast. Science 1996, 273, 622–626. [Google Scholar] [CrossRef] [Green Version]
- Bateman, D.A.; Wickner, R.B. The [PSI+] Prion Exists as a Dynamic Cloud of Variants. PLoS Genet. 2013, 9, e1003257. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, M.; Chien, P.; Naber, N.; Cooke, R.; Weissman, J.S. Conformational Variations in an Infectious Protein Determine Prion Strain Differences. Nature 2004, 428, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Toyama, B.H.; Kelly, M.J.S.; Gross, J.D.; Weissman, J.S. The Structural Basis of Yeast Prion Strain Variants. Nature 2007, 449, 233–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Collins, S.R.; Toyama, B.H.; Weissman, J.S. The Physical Basis of How Prion Conformations Determine Strain Phenotypes. Nature 2006, 442, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Avalos, R.; King, C.-Y.; Wall, J.; Simon, M.; Caspar, D.L.D. Strain-Specific Morphologies of Yeast Prion Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 10165–10170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxa, U.; Cassese, T.; Kajava, A.V.; Steven, A.C. Structure, Function, and Amyloidogenesis of Fungal Prions: Filament Polymorphism and Prion Variants. In Advances in Protein Chemistry; Elsevier: Amsterdam, The Netherlands, 2006; Volume 73, pp. 125–180. ISBN 978-0-12-034273-0. [Google Scholar]
- Kajava, A.V.; Baxa, U.; Wickner, R.B.; Steven, A.C. A Model for Ure2p Prion Filaments and Other Amyloids: The Parallel Superpleated -Structure. Proc. Natl. Acad. Sci. USA 2004, 101, 7885–7890. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, A.K.; MacPhee, C.E.; Zurdo, J.; Morozova-Roche, L.A.; Hill, H.A.O.; Dobson, C.M.; Davis, J.J. Ultrastructural Organization of Amyloid Fibrils ByAtomic Force Microscopy. Biophys. J. 2000, 79, 3282–3293. [Google Scholar] [CrossRef] [Green Version]
- Baxa, U.; Keller, P.W.; Cheng, N.; Wall, J.S.; Steven, A.C. In Sup35p Filaments (the [PSI+] Prion), the Globular C-Terminal Domains Are Widely Offset from the Amyloid Fibril Backbone: Sup35p C-Domains Are Widely Offset from the Amyloid Fibril Backbone. Mol. Microbiol. 2011, 79, 523–532. [Google Scholar] [CrossRef] [Green Version]
- Baxa, U.; Speransky, V.; Steven, A.C.; Wickner, R.B. Mechanism of Inactivation on Prion Conversion of the Saccharomyces cerevisiae Ure2 Protein. Proc. Natl. Acad. Sci. USA 2002, 99, 5253–5260. [Google Scholar] [CrossRef] [Green Version]
- Bousset, L.; Thomson, N.H.; Radford, S.E.; Melki, R. The Yeast Prion Ure2p Retains Its Native α-Helical Conformation upon Assembly into Protein Fibrils in Vitro. EMBO J. 2002, 21, 2903–2911. [Google Scholar] [CrossRef] [Green Version]
- Terry, C.; Wenborn, A.; Gros, N.; Sells, J.; Joiner, S.; Hosszu, L.L.P.; Tattum, M.H.; Panico, S.; Clare, D.K.; Collinge, J.; et al. Ex Vivo Mammalian Prions Are Formed of Paired Double Helical Prion Protein Fibrils. Open Biol. 2016, 6, 160035. [Google Scholar] [CrossRef] [Green Version]
- Castilla, J.; Saá, P.; Hetz, C.; Soto, C. In Vitro Generation of Infectious Scrapie Prions. Cell 2005, 121, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deleault, N.R.; Piro, J.R.; Walsh, D.J.; Wang, F.; Ma, J.; Geoghegan, J.C.; Supattapone, S. Isolation of Phosphatidylethanolamine as a Solitary Cofactor for Prion Formation in the Absence of Nucleic Acids. Proc. Natl. Acad. Sci. USA 2012, 109, 8546–8551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Wang, X.; Yuan, C.-G.; Ma, J. Generating a Prion with Bacterially Expressed Recombinant Prion Protein. Science 2010, 327, 1132–1135. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, C.; Fizet, J.; Properzi, F.; Batchelor, M.; Sandberg, M.K.; Edgeworth, J.A.; Afran, L.; Ho, S.; Badhan, A.; Klier, S.; et al. A Systematic Investigation of Production of Synthetic Prions from Recombinant Prion Protein. Open Biol. 2015, 5, 150165. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; McGovern, G.; Zhang, Y.; Wang, F.; Zha, L.; Jeffrey, M.; Ma, J. Intraperitoneal Infection of Wild-Type Mice with Synthetically Generated Mammalian Prion. PLOS Pathog. 2015, 11, e1004958. [Google Scholar] [CrossRef] [Green Version]
- Pan, C.; Yang, J.; Zhang, X.; Chen, Y.; Wei, S.; Yu, G.; Pan, Y.-H.; Ma, J.; Yuan, C. Oral Ingestion of Synthetically Generated Recombinant Prion Is Sufficient to Cause Prion Disease in Wild-Type Mice. Pathogens 2020, 9, 653. [Google Scholar] [CrossRef]
- Piro, J.R.; Wang, F.; Walsh, D.J.; Rees, J.R.; Ma, J.; Supattapone, S. Seeding Specificity and Ultrastructural Characteristics of Infectious Recombinant Prions. Biochemistry 2011, 50, 7111–7116. [Google Scholar] [CrossRef] [Green Version]
- Terry, C.; Harniman, R.L.; Sells, J.; Wenborn, A.; Joiner, S.; Saibil, H.R.; Miles, M.J.; Collinge, J.; Wadsworth, J.D.F. Structural Features Distinguishing Infectious Ex Vivo Mammalian Prions from Non-Infectious Fibrillar Assemblies Generated in Vitro. Sci. Rep. 2019, 9, 376. [Google Scholar] [CrossRef] [Green Version]
- Schweighauser, M.; Shi, Y.; Tarutani, A.; Kametani, F.; Murzin, A.G.; Ghetti, B.; Matsubara, T.; Tomita, T.; Ando, T.; Hasegawa, K.; et al. Structures of α-Synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469. [Google Scholar] [CrossRef]
- Bansal, A.; Schmidt, M.; Rennegarbe, M.; Haupt, C.; Liberta, F.; Stecher, S.; Puscalau-Girtu, I.; Biedermann, A.; Fändrich, M. AA Amyloid Fibrils from Diseased Tissue Are Structurally Different from in Vitro Formed SAA Fibrils. Nat. Commun. 2021, 12, 1013. [Google Scholar] [CrossRef]
- Annamalai, K.; Liberta, F.; Vielberg, M.-T.; Close, W.; Lilie, H.; Gührs, K.-H.; Schierhorn, A.; Koehler, R.; Schmidt, A.; Haupt, C.; et al. Common Fibril Structures Imply Systemically Conserved Protein Misfolding Pathways In Vivo. Angew. Chem. Int. Ed. 2017, 56, 7510–7514. [Google Scholar] [CrossRef] [PubMed]
- Kollmer, M.; Close, W.; Funk, L.; Rasmussen, J.; Bsoul, A.; Schierhorn, A.; Schmidt, M.; Sigurdson, C.J.; Jucker, M.; Fändrich, M. Cryo-EM Structure and Polymorphism of Aβ Amyloid Fibrils Purified from Alzheimer’s Brain Tissue. Nat. Commun. 2019, 10, 4760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Falcon, B.; Murzin, A.G.; Fan, J.; Crowther, R.A.; Goedert, M.; Scheres, S.H. Heparin-Induced Tau Filaments Are Polymorphic and Differ from Those in Alzheimer’s and Pick’s Diseases. eLife 2019, 8, e43584. [Google Scholar] [CrossRef] [PubMed]
- Glover, J.R.; Kowal, A.S.; Schirmer, E.C.; Patino, M.M.; Liu, J.-J.; Lindquist, S. Self-Seeded Fibers Formed by Sup35, the Protein Determinant of [PSI+], a Heritable Prion-like Factor of S. Cerevisiae. Cell 1997, 89, 811–819. [Google Scholar] [CrossRef] [Green Version]
- King, C.-Y.; Diaz-Avalos, R. Protein-Only Transmission of Three Yeast Prion Strains. Nature 2004, 428, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Pei, F.; DiSalvo, S.; Sindi, S.S.; Serio, T.R. A Dominant-Negative Mutant Inhibits Multiple Prion Variants through a Common Mechanism. PLoS Genet. 2017, 13, e1007085. [Google Scholar] [CrossRef] [Green Version]
- Salnikova, A.B.; Kryndushkin, D.S.; Smirnov, V.N.; Kushnirov, V.V.; Ter-Avanesyan, M.D. Nonsense Suppression in Yeast Cells Overproducing Sup35 (ERF3) Is Caused by Its Non-Heritable Amyloids. J. Biol. Chem. 2005, 280, 8808–8812. [Google Scholar] [CrossRef] [Green Version]
- Gorkovskiy, A.; Reidy, M.; Masison, D.C.; Wickner, R.B. Hsp104 Disaggregase at Normal Levels Cures Many [PSI+] Prion Variants in a Process Promoted by Sti1p, Hsp90, and Sis1p. Proc. Natl. Acad. Sci. USA 2017, 114, E4193–E4202. [Google Scholar] [CrossRef] [Green Version]
- Collinge, J. Prion Strain Mutation and Selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian Evolution of Prions in Cell Culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [Green Version]
- Burke, C.M.; Walsh, D.J.; Steele, A.D.; Agrimi, U.; Di Bari, M.A.; Watts, J.C.; Supattapone, S. Full Restoration of Specific Infectivity and Strain Properties from Pure Mammalian Prion Protein. PLoS Pathog. 2019, 15, e1007662. [Google Scholar] [CrossRef]
- Heerde, T.; Rennegarbe, M.; Biedermann, A.; Savran, D.; Pfeiffer, P.B.; Hitzenberger, M.; Baur, J.; Puscalau-Girtu, I.; Zacharias, M.; Schwierz, N.; et al. Cryo-EM Demonstrates the in Vitro Proliferation of an Ex Vivo Amyloid Fibril Morphology by Seeding. Nat. Commun. 2022, 13, 85. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V. From Posttranslational Modifications to Disease Phenotype: A Substrate Selection Hypothesis in Neurodegenerative Diseases. Int. J. Mol. Sci. 2021, 22, 901. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Baskakov, I.V. Role of Sialylation of N-Linked Glycans in Prion Pathogenesis. Cell Tissue Res. 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.D.; Buxbaum, J.N.; Eisenberg, D.S.; Merlini, G.; Saraiva, M.J.M.; Sekijima, Y.; Sipe, J.D.; Westermark, P. Amyloid Nomenclature 2020: Update and Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2020, 27, 217–222. [Google Scholar] [CrossRef]
- Haas, C.; Steiner, H. Protofibrils, the Unifying Toxic Molecule of Neurodegenerative Disorders? Nat. Neurosci. 2001, 4, 859–860. [Google Scholar] [CrossRef]
- Bigi, A.; Ermini, E.; Chen, S.W.; Cascella, R.; Cecchi, C. Exploring the Release of Toxic Oligomers from α-Synuclein Fibrils with Antibodies and STED Microscopy. Life 2021, 11, 431. [Google Scholar] [CrossRef]
- Benilova, I.; Karran, E.; De Strooper, B. The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci. 2012, 15, 349–357. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Hartley, D.M.; Petre, B.M.; Wall, J.S.; Simon, M.N.; Walz, T.; Lansbury, P.T. Mixtures of Wild-Type and a Pathogenic (E22G) Form of Aβ40 in Vitro Accumulate Protofibrils, Including Amyloid Pores. J. Mol. Biol. 2003, 332, 795–808. [Google Scholar] [CrossRef] [Green Version]
- Laurents, D.V.; Gorman, P.M.; Guo, M.; Rico, M.; Chakrabartty, A.; Bruix, M. Alzheimer’s Aβ40 Studied by NMR at Low PH Reveals That Sodium 4,4-Dimethyl-4-Silapentane-1-Sulfonate (DSS) Binds and Promotes β-Ball Oligomerization. J. Biol. Chem. 2005, 280, 3675–3685. [Google Scholar] [CrossRef] [Green Version]
- Giehm, L.; Svergun, D.I.; Otzen, D.E.; Vestergaard, B. Low-Resolution Structure of a Vesicle Disrupting -Synuclein Oligomer That Accumulates during Fibrillation. Proc. Natl. Acad. Sci. USA 2011, 108, 3246–3251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent Toxicity of Aggregates Implies a Common Mechanism for Protein Misfolding Diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [Green Version]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Vbeyreuther, K.; Bush, A.I.; Masters, C.L. Soluble Pool of Aβ Amyloid as a Determinant of Severity of Neurodegeneration in Alzheimer’s Disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Mc Donald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. The Presence of Sodium Dodecyl Sulphate-Stable Aβ Dimers Is Strongly Associated with Alzheimer-Type Dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Ideno, S.; et al. Isolation and Characterization of Patient-Derived, Toxic, High Mass Amyloid β-Protein (Aβ) Assembly from Alzheimer Disease Brains. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef] [Green Version]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-β Protein Dimers Isolated Directly from Alzheimer’s Brains Impair Synaptic Plasticity and Memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid-Protein Dimers Rapidly Form Stable Synaptotoxic Protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef]
- Sajnani, G.; Silva, C.J.; Ramos, A.; Pastrana, M.A.; Onisko, B.C.; Erickson, M.L.; Antaki, E.M.; Dynin, I.; Vázquez-Fernández, E.; Sigurdson, C.J.; et al. PK-Sensitive PrPSc Is Infectious and Shares Basic Structural Features with PK-Resistant PrPSc. PLoS Pathog. 2012, 8, e1002547. [Google Scholar] [CrossRef] [Green Version]
- Silveira, J.R.; Raymond, G.J.; Hughson, A.G.; Race, R.E.; Sim, V.L.; Hayes, S.F.; Caughey, B. The Most Infectious Prion Protein Particles. Nature 2005, 437, 257–261. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, J.L.; Nettleton, E.J.; Bouchard, M.; Robinson, C.V.; Dobson, C.M.; Saibil, H.R. The Protofilament Structure of Insulin Amyloid Fibrils. Proc. Natl. Acad. Sci. USA 2002, 99, 9196–9201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, R.; Ionescu-Zanetti, C.; Pope, M.; Li, J.; Nielson, L.; Ramírez-Alvarado, M.; Regan, L.; Fink, A.L.; Carter, S.A. A General Model for Amyloid Fibril Assembly Based on Morphological Studies Using Atomic Force Microscopy. Biophys. J. 2003, 85, 1135–1144. [Google Scholar] [CrossRef] [Green Version]
- Serpell, L.C.; Sunde, M.; Benson, M.D.; Tennent, G.A.; Pepys, M.B.; Fraser, P.E. The Protofilament Substructure of Amyloid Fibrils11Edited by F. E. Cohen. J. Mol. Biol. 2000, 300, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Liberta, F.; Loerch, S.; Rennegarbe, M.; Schierhorn, A.; Westermark, P.; Westermark, G.T.; Hazenberg, B.P.C.; Grigorieff, N.; Fändrich, M.; Schmidt, M. Cryo-EM Fibril Structures from Systemic AA Amyloidosis Reveal the Species Complementarity of Pathological Amyloids. Nat. Commun. 2019, 10, 1104. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Vázquez-Fernández, E.; Vos, M.R.; Afanasyev, P.; Cebey, L.; Sevillano, A.M.; Vidal, E.; Rosa, I.; Renault, L.; Ramos, A.; Peters, P.J.; et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog. 2016, 12, e1005835. [Google Scholar] [CrossRef]
- Groveman, B.R.; Dolan, M.A.; Taubner, L.M.; Kraus, A.; Wickner, R.B.; Caughey, B. Parallel In-Register Intermolecular β-Sheet Architectures for Prion-Seeded Prion Protein (PrP) Amyloids. J. Biol. Chem. 2014, 289, 24129–24142. [Google Scholar] [CrossRef] [Green Version]
- Kraus, A.; Hoyt, F.; Schwartz, C.L.; Hansen, B.; Artikis, E.; Hughson, A.G.; Raymond, G.J.; Race, B.; Baron, G.S.; Caughey, B. High-Resolution Structure and Strain Comparison of Infectious Mammalian Prions. Mol. Cell 2021, 81, 4540–4551.e6. [Google Scholar] [CrossRef]
- Radamaker, L.; Baur, J.; Huhn, S.; Haupt, C.; Hegenbart, U.; Schönland, S.; Bansal, A.; Schmidt, M.; Fändrich, M. Cryo-EM Reveals Structural Breaks in a Patient-Derived Amyloid Fibril from Systemic AL Amyloidosis. Nat. Commun. 2021, 12, 875. [Google Scholar] [CrossRef]
- Wenborn, A.; Terry, C.; Gros, N.; Joiner, S.; D’Castro, L.; Panico, S.; Sells, J.; Cronier, S.; Linehan, J.M.; Brandner, S.; et al. A Novel and Rapid Method for Obtaining High Titre Intact Prion Strains from Mammalian Brain. Sci. Rep. 2015, 5, 10062. [Google Scholar] [CrossRef]
- Serio, T.R.; Cashikar, A.G.; Kowal, A.S.; Sawicki, G.J.; Moslehi, J.J.; Serpell, L.; Arnsdorf, M.F.; Lindquist, S.L. Nucleated Conformational Conversion and the Replication of Conformational Information by a Prion Determinant. Science 2000, 289, 1317–1321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagriantsev, S.; Liebman, S.W. Specificity of Prion Assembly in Vivo. J. Biol. Chem. 2004, 279, 51042–51048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derdowski, A.; Sindi, S.S.; Klaips, C.L.; DiSalvo, S.; Serio, T.R. A Size Threshold Limits Prion Transmission and Establishes Phenotypic Diversity. Science 2010, 330, 680–683. [Google Scholar] [CrossRef] [Green Version]
- Pezza, J.A.; Villali, J.; Sindi, S.S.; Serio, T.R. Amyloid-Associated Activity Contributes to the Severity and Toxicity of a Prion Phenotype. Nat. Commun. 2014, 5, 4384. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, A.V.; Belashova, T.A.; Bondarev, S.A.; Velizhanina, M.E.; Barbitoff, Y.A.; Matveenko, A.G.; Valina, A.A.; Simanova, A.L.; Zhouravleva, G.A.; Galkin, A.P. Direct Proof of the Amyloid Nature of Yeast Prions [PSI+] and [PIN+] by the Method of Immunoprecipitation of Native Fibrils. FEMS Yeast Res. 2021, 21, foab046. [Google Scholar] [CrossRef] [PubMed]
- Speransky, V.V.; Taylor, K.L.; Edskes, H.K.; Wickner, R.B.; Steven, A.C. Prion Filament Networks in [Ure3] Cells of Saccharomyces Cerevisiae. J. Cell Biol. 2001, 153, 1327–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coustou-Linares, V.; Maddelein, M.-L.; Bégueret, J.; Saupe, S.J. In Vivo Aggregation of the HET-s Prion Protein of the Fungus Podospora Anserina: In Vivo Aggregation of the HET-s Prion Protein of P. Anserina. Mol. Microbiol. 2002, 42, 1325–1335. [Google Scholar] [CrossRef]
- Edskes, H.K.; Gray, V.T.; Wickner, R.B. The [URE3] Prion Is an Aggregated Form of Ure2p That Can Be Cured by Overexpression of Ure2p Fragments. Proc. Natl. Acad. Sci. USA 1999, 96, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Sondheimer, N.; Lindquist, S. Rnq1: An Epigenetic Modifier of Protein Function in Yeast. Mol. Cell 2000, 5, 163–172. [Google Scholar] [CrossRef]
- Satpute-Krishnan, P.; Langseth, S.X.; Serio, T.R. Hsp104-Dependent Remodeling of Prion Complexes Mediates Protein-Only Inheritance. PLoS Biol. 2007, 5, e24. [Google Scholar] [CrossRef] [Green Version]
- Satpute-Krishnan, P.; Serio, T.R. Prion Protein Remodelling Confers an Immediate Phenotypic Switch. Nature 2005, 437, 262–265. [Google Scholar] [CrossRef]
- Kimura, Y.; Koitabashi, S.; Fujita, T. Analysis of Yeast Prion Aggregates with Amyloid-Staining Compound In Vivo. Cell Struct. Funct. 2003, 28, 187–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkatch, I.L.; Bradley, M.E.; Hong, J.Y.; Liebman, S.W. Prions Affect the Appearance of Other Prions: The Story of [PIN+]. Cell 2001, 106, 171–182. [Google Scholar] [CrossRef] [Green Version]
- Osherovich, L.Z.; Weissman, J.S. Multiple Gln/Asn-Rich Prion Domains Confer Susceptibility to Induction of the Yeast [PSI] Prion. Cell 2001, 106, 183–194. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Derkatch, I.L.; Liebman, S.W. The Relationship between Visible Intracellular Aggregates That Appear after Overexpression of Sup35 and the Yeast Prion-like Elements [PSI+] and [PIN+]. Mol. Microbiol. 2001, 39, 37–46. [Google Scholar] [CrossRef]
- Taneja, V.; Maddelein, M.-L.; Talarek, N.; Saupe, S.J.; Liebman, S.W. A Non-Q/N-Rich Prion Domain of a Foreign Prion, [Het-s], Can Propagate as a Prion in Yeast. Mol. Cell 2007, 27, 67–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathur, V.; Taneja, V.; Sun, Y.; Liebman, S.W. Analyzing the Birth and Propagation of Two Distinct Prions, PSI+ and [Het-s], in Yeast. Mol. Biol. Cell 2010, 21, 1449–1461. [Google Scholar] [CrossRef] [Green Version]
- Arslan, F.; Hong, J.Y.; Kanneganti, V.; Park, S.-K.; Liebman, S.W. Heterologous Aggregates Promote De Novo Prion Appearance via More than One Mechanism. PLoS Genet. 2015, 11, e1004814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Wisniewski, B.T.; Paulson, E.; Obaoye, J.O.; Merrill, S.J.; Manogaran, A.L. De Novo [PSI+] Prion Formation Involves Multiple Pathways to Form Infectious Oligomers. Sci. Rep. 2017, 7, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaganovich, D.; Kopito, R.; Frydman, J. Misfolded Proteins Partition between Two Distinct Quality Control Compartments. Nature 2008, 454, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Kabani, M.; Melki, R. Sup35p in Its Soluble and Prion States Is Packaged inside Extracellular Vesicles. mBio 2015, 6, e01017-15. [Google Scholar] [CrossRef] [Green Version]
- Kabani, M. Extracellular Vesicles-Encapsulated Yeast Prions and What They Can Tell Us about the Physical Nature of Propagons. Int. J. Mol. Sci. 2020, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Ganusova, E.E.; Ozolins, L.N.; Bhagat, S.; Newnam, G.P.; Wegrzyn, R.D.; Sherman, M.Y.; Chernoff, Y.O. Modulation of Prion Formation, Aggregation, and Toxicity by the Actin Cytoskeleton in Yeast. Mol. Cell. Biol. 2006, 26, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailleul-Winslett, P.A.; Newnam, G.P.; Wegrzyn, R.D.; Chernoff, Y.O. An Antiprion Effect of the Anticytoskeletal Drug Latrunculin A in Yeast. Gene Expr. 2001, 9, 145–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbinden, A.; Pérez-Berlanga, M.; De Rossi, P.; Polymenidou, M. Phase Separation and Neurodegenerative Diseases: A Disturbance in the Force. Dev. Cell 2020, 55, 45–68. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Mukhopadhyay, S. Prion Protein Biology Through the Lens of Liquid-Liquid Phase Separation. J. Mol. Biol. 2022, 434, 167368. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Krishnan, R.; Lemke, E.A.; Lindquist, S.; Deniz, A.A. A Natively Unfolded Yeast Prion Monomer Adopts an Ensemble of Collapsed and Rapidly Fluctuating Structures. Proc. Natl. Acad. Sci. USA 2007, 104, 2649–2654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnan, R.; Goodman, J.L.; Mukhopadhyay, S.; Pacheco, C.D.; Lemke, E.A.; Deniz, A.A.; Lindquist, S. Conserved Features of Intermediates in Amyloid Assembly Determine Their Benign or Toxic States. Proc. Natl. Acad. Sci. USA 2012, 109, 11172–11177. [Google Scholar] [CrossRef] [Green Version]
- Franzmann, T.M.; Jahnel, M.; Pozniakovsky, A.; Mahamid, J.; Holehouse, A.S.; Nüske, E.; Richter, D.; Baumeister, W.; Grill, S.W.; Pappu, R.V.; et al. Phase Separation of a Yeast Prion Protein Promotes Cellular Fitness. Science 2018, 359, eaao5654. [Google Scholar] [CrossRef] [Green Version]
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular Condensates: Organizers of Cellular Biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298. [Google Scholar] [CrossRef]
- Gao, Z.; Zhang, W.; Chang, R.; Zhang, S.; Yang, G.; Zhao, G. Liquid-Liquid Phase Separation: Unraveling the Enigma of Biomolecular Condensates in Microbial Cells. Front. Microbiol. 2021, 12, 751880. [Google Scholar] [CrossRef] [PubMed]
- Manogaran, A.L.; Hong, J.Y.; Hufana, J.; Tyedmers, J.; Lindquist, S.; Liebman, S.W. Prion Formation and Polyglutamine Aggregation Are Controlled by Two Classes of Genes. PLoS Genet. 2011, 7, e1001386. [Google Scholar] [CrossRef] [Green Version]
- Cox, B.; Ness, F.; Tuite, M. Analysis of the Generation and Segregation of Propagons: Entities That Propagate the [PSI+] Prion in Yeast. Genetics 2003, 165, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Ness, F.; Ferreira, P.; Cox, B.S.; Tuite, M.F. Guanidine Hydrochloride Inhibits the Generation of Prion “Seeds” but Not Prion Protein Aggregation in Yeast. Mol. Cell Biol. 2002, 22, 5593–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai-Noma, S.; Ayano, S.; Pack, C.-G.; Kinjo, M.; Yoshida, M.; Yasuda, K.; Taguchi, H. Dynamics of Yeast Prion Aggregates in Single Living Cells. Genes Cells 2006, 11, 1085–1096. [Google Scholar] [CrossRef] [PubMed]
- Chernoff, Y.O.; Lindquist, S.L.; Bun-ichiro, O.; Inge-Vechtomov, S.G.; Liebman, S.W. Role of the Chaperone Protein Hsp104 in Propagation of the Yeast Prion-Like Factor [Psi+]. Science 1995, 268, 880–884. [Google Scholar] [CrossRef]
- Wegrzyn, R.D.; Bapat, K.; Newnam, G.P.; Zink, A.D.; Chernoff, Y.O. Mechanism of Prion Loss after Hsp104 Inactivation in Yeast. Mol. Cell. Biol. 2001, 21, 4656–4669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai-Noma, S.; Pack, C.-G.; Tsuji, T.; Kinjo, M.; Taguchi, H. Single Mother-Daughter Pair Analysis to Clarify the Diffusion Properties of Yeast Prion Sup35 in Guanidine-HCl-Treated [PSI+] Cells. Genes Cells 2009, 14, 1045–1054. [Google Scholar] [CrossRef]
- Borchsenius, A.S.; Müller, S.; Newnam, G.P.; Inge-Vechtomov, S.G.; Chernoff, Y.O. Prion Variant Maintained Only at High Levels of the Hsp104 Disaggregase. Curr. Genet. 2006, 49, 21–29. [Google Scholar] [CrossRef]
- Kushnirov, V.V.; Kochneva-Pervukhova, N.V.; Chechenova, M.B.; Frolova, N.S.; Ter-Avanesyan, M.D. Prion Properties of the Sup35 Protein of Yeast Pichia Methanolica. EMBO J. 2000, 19, 324–331. [Google Scholar] [CrossRef] [Green Version]
- Winkler, J.; Tyedmers, J.; Bukau, B.; Mogk, A. Hsp70 Targets Hsp100 Chaperones to Substrates for Protein Disaggregation and Prion Fragmentation. J. Cell Biol. 2012, 198, 387–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ness, F.; Cox, B.S.; Wongwigkarn, J.; Naeimi, W.R.; Tuite, M.F. Over-Expression of the Molecular Chaperone Hsp104 in Saccharomyces cerevisiae Results in the Malpartition of [PSI+] Propagons: Prion Curing by Hsp104 over-Expression. Mol. Microbiol. 2017, 104, 125–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-N.; Morales, D.; Rubinson, E.H.; Masison, D.; Eisenberg, E.; Greene, L.E. Differences in the Curing of [PSI+] Prion by Various Methods of Hsp104 Inactivation. PLoS ONE 2012, 7, e37692. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Rodriguez, R.; Silberman, R.E.; Ahearn, J.M.; Saidha, S.; Cummins, K.C.; Eisenberg, E.; Greene, L.E. Heat Shock Protein 104 (Hsp104)-Mediated Curing of [PSI+] Yeast Prions Depends on Both [PSI+] Conformation and the Properties of the Hsp104 Homologs. J. Biol. Chem. 2017, 292, 8630–8641. [Google Scholar] [CrossRef] [Green Version]
- Jung, G.; Jones, G.; Wegrzyn, R.D.; Masison, D.C. A Role for Cytosolic Hsp70 in Yeast [PSI+] Prion Propagation and [PSI+] as a Cellular Stress. Genetics 2000, 156, 559–570. [Google Scholar] [CrossRef]
- Newnam, G.P.; Wegrzyn, R.D.; Lindquist, S.L.; Chernoff, Y.O. Antagonistic Interactions between Yeast Chaperones Hsp104 and Hsp70 in Prion Curing. Mol. Cell. Biol. 1999, 19, 1325–1333. [Google Scholar] [CrossRef] [Green Version]




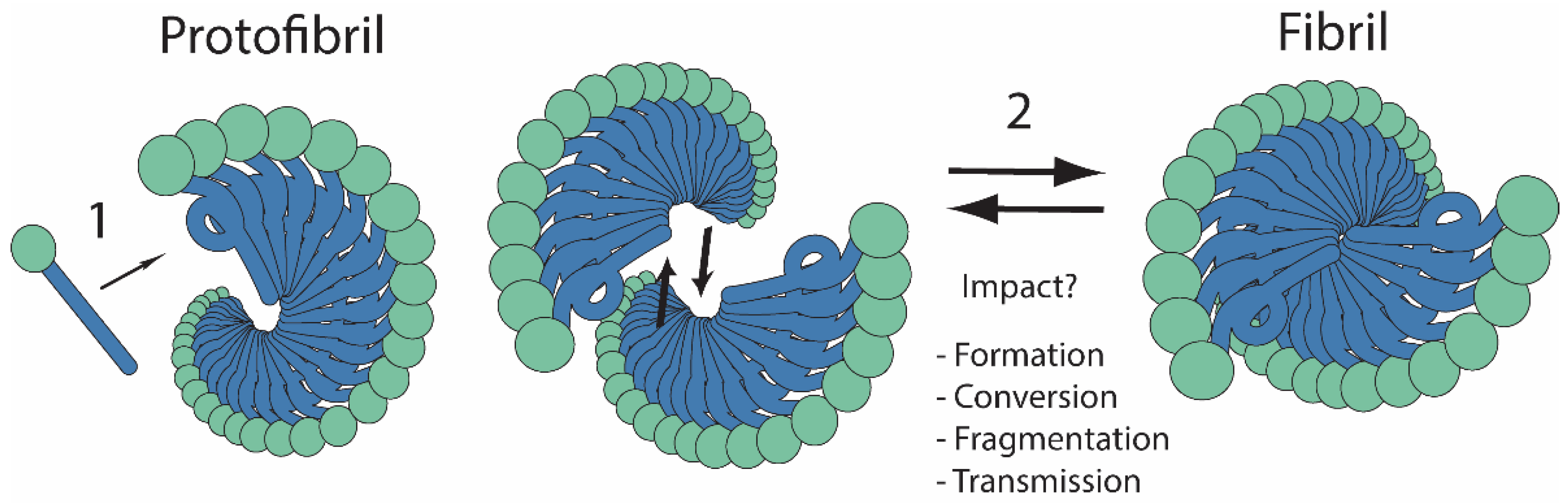{kind=link}
{kind=link}
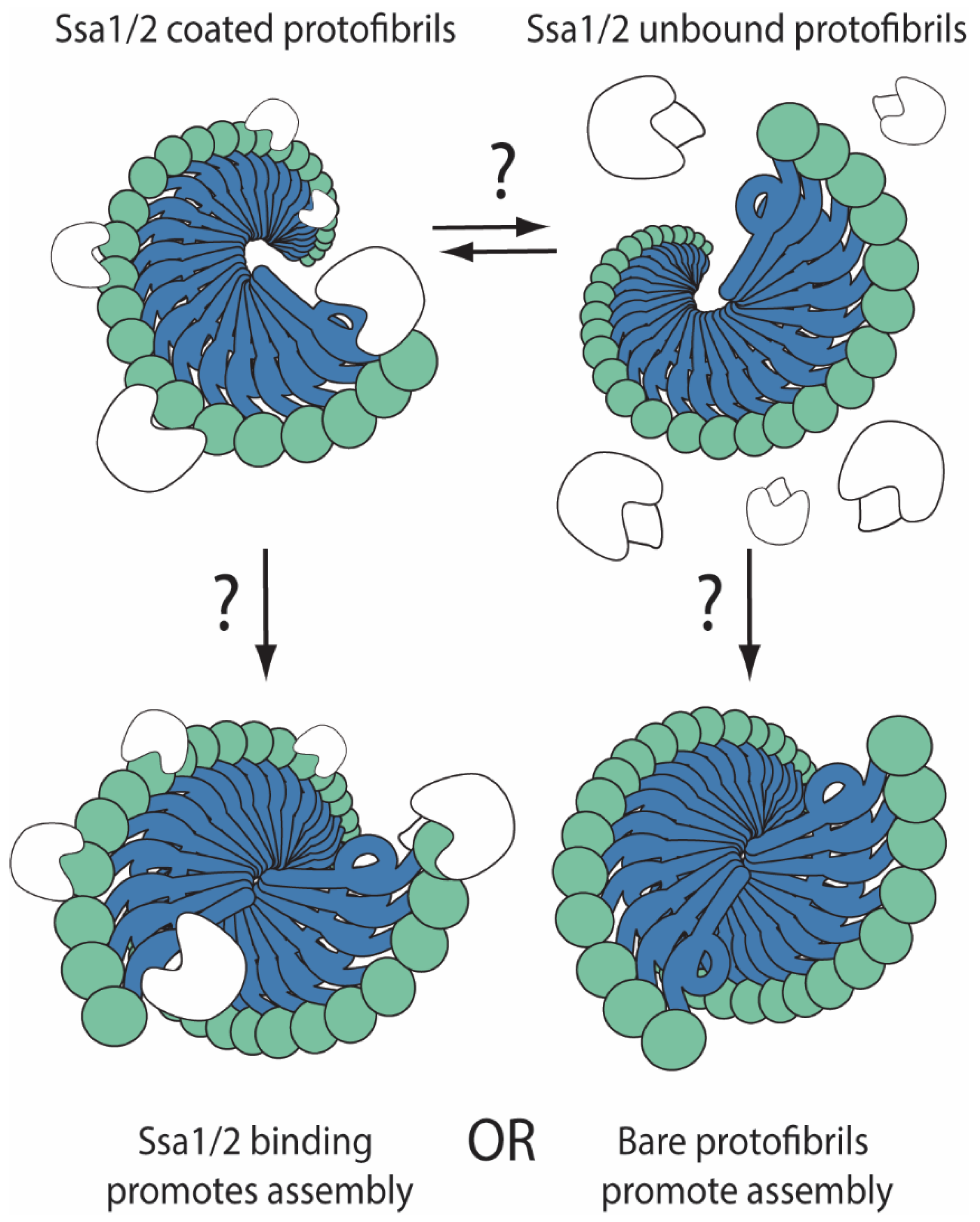{kind=link}
| Amyloid Source | Disruption Method | Extraction Type | Enzymatic Digestion | Detergent | Max. Sediment | Observed Fibril Morphology By EM | Reference |
|---|---|---|---|---|---|---|---|
| Prion Disease–PrPSc | |||||||
| RML PrPSc fibrils | Homogenized (tissue grinder) | Selective protein precipitation: NaPTA | Pronase Benzonase | Sarkosyl | 16,100× g | 2 protofibrils per fibril | [49] |
| GPI-anchorless PrP (27-30) RML PrPSc fibrils from mouse brain | Dounce homogenization High salt | Detergent resistant membrane (DRM): Brij-96 and high salt: NaCl extraction | Benzonase PK | Brij-96 Sarkosyl | Approx. 200,000× g | 2 protofibrils per fibril | [87] |
| aRML fibrils from mouse brain or 263K PrPSc from hamster brain | Dounce homogenization Sonicated | Water | PK | Sulfobetaine | 225,000× g | 1 protofibril per fibil PRIBS-based model for PrPSc in which a single PrP molecule spans the entire fibril width to give an asymmetric cross-section | [89] |
| AL Amyloidosis–Amyloid Light-Chain (AL) | |||||||
| AL amyloidosis human heart tissue | Kontes pellet pestle | Water | Collagenase | no | 3100× g | i and ii polymorphism types by fibril width, pitch and width of crossover | [52] |
| Systemic AL amyloidosis human heart tissue | Kontes pellet pestle | Water | Collagenase | no | 3100× g | 1 protofibril per fibril | [90] |
| AA Amyloidosis–Serum Amyloid A (SAA) | |||||||
| AA amyloid fibrils from human kidney | Kontes pellet pestle | Water | Collagenase | no | 3100× g | 2 protofibrils per fibril | [85] |
| AA amyloid fibrils from mouse liver | Homogenized with scalpel | Water | Collagenase | no | 3100× g | 2–3 protofibrils per fibril | [51,63] |
| Multiple System Atrophy (MSA)–α-syn | |||||||
| α-syn fibrils from MSA human brain | Homogenized | Sarkosyl-insolubility | no | Sarkosyl | 166,000× g | 2 protofibrils per fibril | [50] |
| Alzheimer’s Disease (AD)–Aβ and tau | |||||||
| Aβ fibrils from human AD brain | Homogenized (scalpel) | Water | Collagenase | no | 12,000× g | 1–3 protofibrils per fibril | [53] |
| Tau fibrils from human AD brain | Homogenized (polytron) | Sarkosyl-insolubility Superose 6 increase column Vivacon 500 conc. | Pronase | Sarkosyl | 100,000× g | 2 protofibrils per fibril Paired helical filaments (PHFs) and straight filaments (SFs) each composed of common protofibril structure | [86] |
| Hsp104 | Sup35 | Curing | Time Point Post Induction at Which Observations Made | In Vivo Observation | Ex Vivo Observations (Native) | Ex Vivo Observations (Detergent Present) | Reference |
|---|---|---|---|---|---|---|---|
| Multi copy plasmid | WT | ? | Stable expression | ? | Size shift to smaller fractions when sedimented through 30% sucrose | ? | [131] |
| GAL inducible plasmid for 48 h | NM-Gfp driven from CUP inducible plasmid for 48 h | ✓ | 48 h | Ring aggregate patterns | ? | ? | [106] |
| Multi copy plasmid | WT | ? | Stable expression | ? | ? | Size shift larger we fractionated by SDD-AGE. Increased monomer signal | [9] |
| CUP inducible plasmid for 8 h | NM-Yfp driven from GAL inducible plasmid for 10 h | ? | 10 h | Sup35NM-Yfp les mobile as determined by FLIP and localized to the cytoplasm | ? | ? | [132] |
| GAL inducible plasmid | WT | ✓ | 4 generations | Propagons malpartition to mothers | Soluble pool increase | No size shift when fractionated by SD-AGE | [133] |
| GAL inducible plasmid TET inducible plasmid | Sup35N-Gfp-MC | GAL ✓ TET X | GAL 1 generation TET 12 generations | Loss of detectable foci that are recovered by 1 h water treat | ? | ? | [135] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naeimi, W.R.; Serio, T.R. Beyond Amyloid Fibers: Accumulation, Biological Relevance, and Regulation of Higher-Order Prion Architectures. Viruses 2022, 14, 1635. https://doi.org/10.3390/v14081635
Naeimi WR, Serio TR. Beyond Amyloid Fibers: Accumulation, Biological Relevance, and Regulation of Higher-Order Prion Architectures. Viruses. 2022; 14(8):1635. https://doi.org/10.3390/v14081635
Chicago/Turabian StyleNaeimi, Wesley R., and Tricia R. Serio. 2022. "Beyond Amyloid Fibers: Accumulation, Biological Relevance, and Regulation of Higher-Order Prion Architectures" Viruses 14, no. 8: 1635. https://doi.org/10.3390/v14081635