
Is the HTLV-1 Retrovirus Targeted by Host Restriction Factors?


Abstract
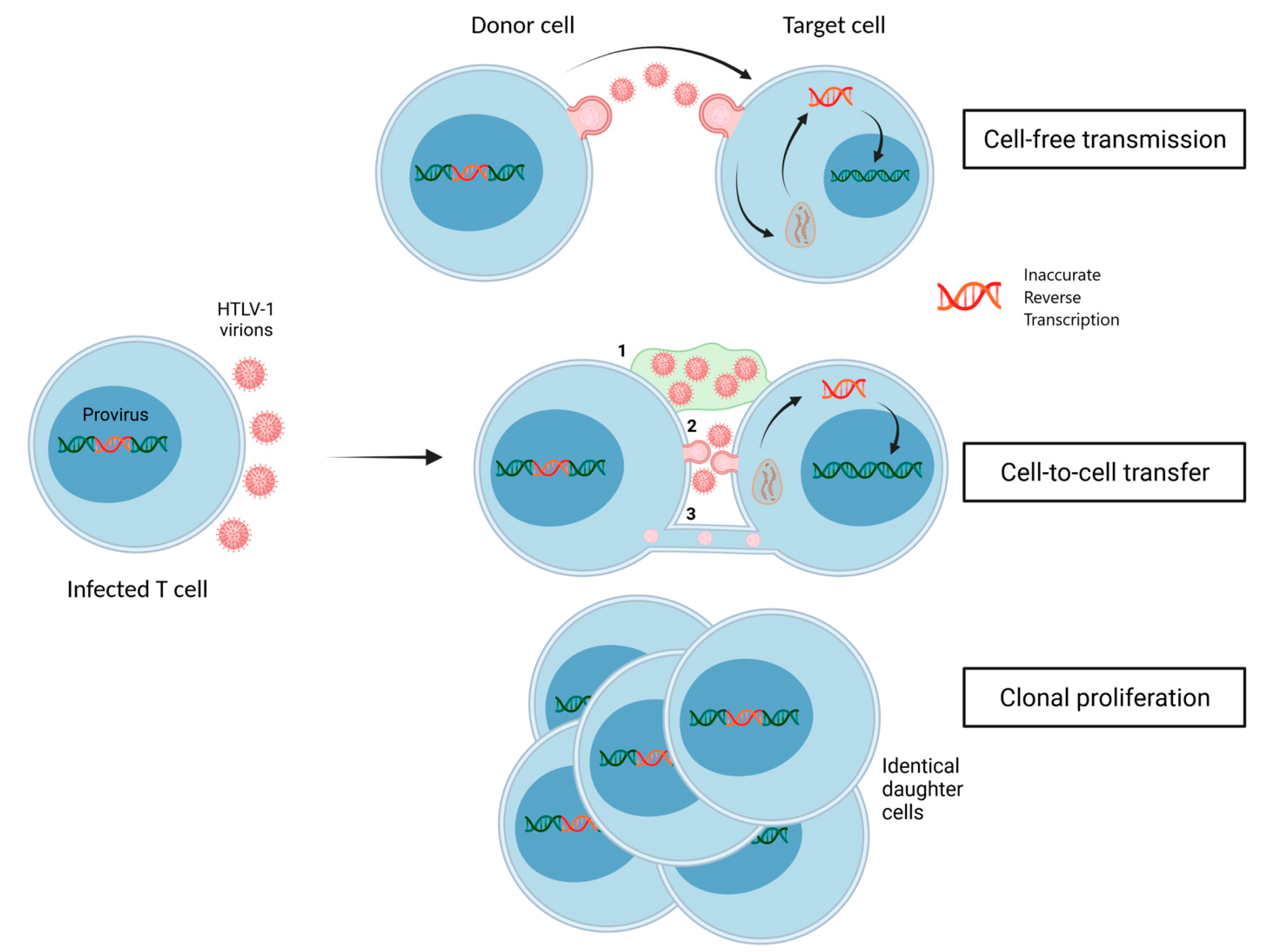
:1. Introduction
2. HTLV-1 Is Poorly Sensitive to Type-I Interferon, a Potent Inducer of HIV Restriction Factors
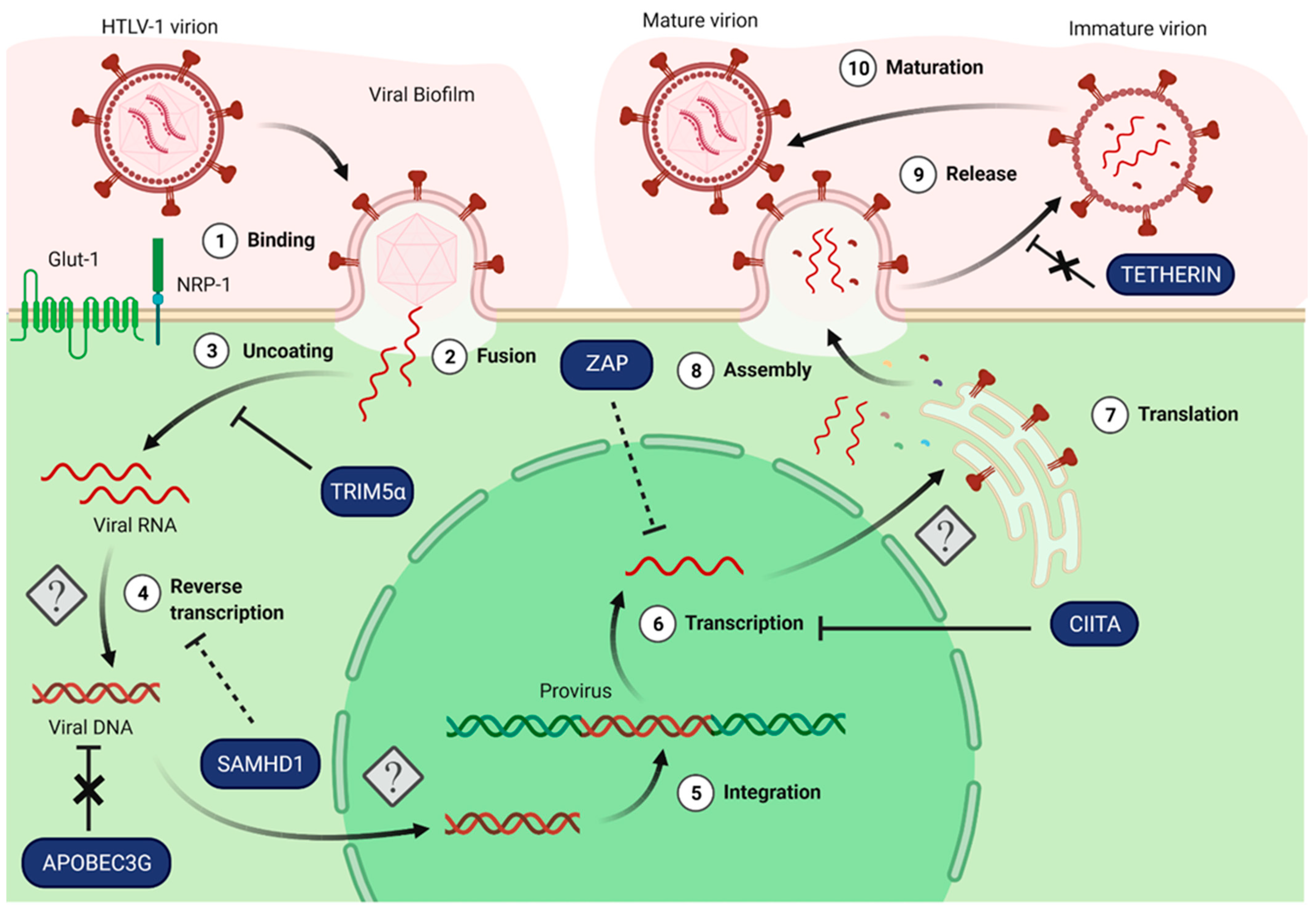
3. Activity of Known HIV Restriction Factors on HTLV-1 Replication
3.1. Factors Targeting Viral Uncoating: TRIM5α
3.2. Factors Targeting Viral DNA Synthesis: APOBEC3 and SAMHD1
3.3. Factors Targeting Viral Expression: CIITA and ZAP
3.4. Factors Targeting Viral Release and Viral Spread: Tetherin
4. How Can We Further Expand Our Understanding of HTLV-1 Restriction?
4.1. By Increasing Our Knowledge on HTLV-1 Sensing and Induction of Restriction Factors upon Dendritic Cell Maturation
4.2. By Interrogating the Possible Relevance of Other Known Restriction Factors Active against HIV or Other Retroviruses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gessain, A.; Cassar, O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gessain, A.; Barin, F.; Vernant, J.C.; Gout, O.; Maurs, L.; Calender, A.; de Thé, G. Antibodies to Human T-Lymphotropic Virus Type-I in Patients with Tropical Spastic Paraparesis. Lancet 1985, 2, 407–410. [Google Scholar] [CrossRef]
- Uchiyama, T.; Yodoi, J.; Sagawa, K.; Takatsuki, K.; Uchino, H. Adult T-Cell Leukemia: Clinical and Hematologic Features of 16 Cases. Blood 1977, 50, 481–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eusebio-Ponce, E.; Anguita, E.; Paulino-Ramirez, R.; Javier Candel, F. HTLV-1 Infection: An Emerging Risk. Pathogenesis, Epidemiology, Diagnosis and Associated Diseases. Rev. Esp. Quim. 2019, 32, 485–496. [Google Scholar]
- Rocamonde, B.; Carcone, A.; Mahieux, R.; Dutartre, H. HTLV-1 Infection of Myeloid Cells: From Transmission to Immune Alterations. Retrovirology 2019, 16, 45. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.S.; Petrow-Sadowski, C.; Huang, Y.K.; Bertolette, D.C.; Ruscetti, F.W. Cell-Free HTLV-1 Infects Dendritic Cells Leading to Transmission and Transformation of CD4+ T Cells. Nat. Med. 2008, 14, 429–436. [Google Scholar] [CrossRef]
- Alais, S.; Mahieux, R.; Dutartre, H. Viral Source-Independent High Susceptibility of Dendritic Cells to Human T-Cell Leukemia Virus Type 1 Infection Compared to That of T Lymphocytes. J. Virol. 2015, 89, 10580–10590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizkallah, G.; Alais, S.; Futsch, N.; Tanaka, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Dendritic Cell Maturation, but Not Type I Interferon Exposure, Restricts Infection by HTLV-1, and Viral Transmission to T-Cells. PLoS Pathog. 2017, 13, e1006353. [Google Scholar] [CrossRef]
- Wattel, E.; Cavrois, M.; Gessain, A.; Wain-Hobson, S. Clonal Expansion of Infected Cells: A Way of Life for HTLV-I. J. Acquir. Immune Defic. Syndr. 1996, 13, S92–S99. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Mateus, M.; Thinnes, C.C.; McCullagh, J.S.; Schofield, C.J.; Taylor, G.P.; Bangham, C.R.M. Glucose Metabolism and Oxygen Availability Govern Reactivation of the Latent Human Retrovirus HTLV-1. Cell Chem. Biol. 2017, 24, 1377–1387.e3. [Google Scholar] [CrossRef] [Green Version]
- Miura, M.; Dey, S.; Ramanayake, S.; Singh, A.; Rueda, D.S.; Bangham, C.R.M. Kinetics of HTLV-1 Reactivation from Latency Quantified by Single-Molecule RNA FISH and Stochastic Modelling. PLOS Pathog. 2019, 15, e1008164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derse, D.; Hill, S.A.; Lloyd, P.A.; Chung, H.K.; Morse, B.A. Examining Human T-Lymphotropic Virus Type 1 Infection and Replication by Cell-Free Infection with Recombinant Virus Vectors. J. Virol. 2001, 75, 8461–8468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derse, D.; Heidecker, G.; Mitchell, M.; Hill, S.; Lloyd, P.; Princler, G. Infectious Transmission and Replication of Human T-Cell Leukemia Virus Type 1. Front. Biosci. 2004, 9, 2495–2499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igakura, T.; Stinchcombe, J.C.; Goon, P.K.C.; Taylor, G.P.; Weber, J.N.; Griffiths, G.M.; Tanaka, Y.; Osame, M.; Bangham, C.R.M. Spread of HTLV-I between Lymphocytes by Virus-Induced Polarization of the Cytoskeleton. Science 2003, 299, 1713–1716. [Google Scholar] [CrossRef] [Green Version]
- Fan, N.; Gavalchin, J.; Paul, B.; Wells, K.H.; Lane, M.J.; Poiesz, B.J. Infection of Peripheral Blood Mononuclear Cells and Cell Lines by Cell-Free Human T-Cell Lymphoma/Leukemia Virus Type I. J. Clin. Microbiol. 1992, 30, 905–910. [Google Scholar] [CrossRef] [Green Version]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef] [Green Version]
- Malim, M.H.; Bieniasz, P.D. HIV Restriction Factors and Mechanisms of Evasion. Cold Spring Harb. Perspect. Med. 2012, 2, a006940. [Google Scholar] [CrossRef]
- Doyle, T.; Goujon, C.; Malim, M.H. HIV-1 and Interferons: Who’s Interfering with Whom? Nat. Rev. Microbiol. 2015, 13, 403–413. [Google Scholar] [CrossRef]
- Kluge, S.F.; Sauter, D.; Kirchhoff, F. SnapShot: Antiviral Restriction Factors. Cell 2015, 163, 774. [Google Scholar] [CrossRef] [Green Version]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx Relieves Inhibition of HIV-1 Infection of Macrophages Mediated by the SAMHD1 Protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef] [Green Version]
- Van Damme, N.; Goff, D.; Katsura, C.; Jorgenson, R.L.; Mitchell, R.; Johnson, M.C.; Stephens, E.B.; Guatelli, J. The Interferon-Induced Protein BST-2 Restricts HIV-1 Release and Is Downregulated from the Cell Surface by the Viral Vpu Protein. Cell Host Microbe 2008, 3, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinpara, S.; Hasegawa, A.; Utsunomiya, A.; Nishitsuji, H.; Furukawa, H.; Masuda, T.; Kannagi, M. Stromal Cell-Mediated Suppression of Human T-Cell Leukemia Virus Type 1 Expression In Vitro and In Vivo by Type I Interferon. J. Virol. 2009, 83, 5101–5108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinpara, S.; Kijiyama, M.; Takamori, A.; Hasegawa, A.; Sasada, A.; Masuda, T.; Tanaka, Y.; Utsunomiya, A.; Kannagi, M. Interferon-α (IFN-α) Suppresses HTLV-1 Gene Expression and Cell Cycling, While IFN-α Combined with Zidovudine Induces P53 Signaling and Apoptosis in HTLV-1-Infected Cells. Retrovirology 2013, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khouri, R.; Silva-Santos, G.; Dierckx, T.; Menezes, S.M.; Decanine, D.; Theys, K.; Silva, A.C.; Farré, L.; Bittencourt, A.; Mangino, M.; et al. A Genetic IFN/STAT1/FAS Axis Determines CD4 T Stem Cell Memory Levels and Apoptosis in Healthy Controls and Adult T-Cell Leukemia Patients. Oncoimmunology 2018, 7, e1426423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cachat, A.; Chevalier, S.A.; Alais, S.; Ko, N.L.; Ratner, L.; Journo, C.; Dutartre, H.; Mahieux, R. Alpha Interferon Restricts Human T-Lymphotropic Virus Type 1 and 2 de Novo Infection through PKR Activation. J. Virol. 2013, 87, 13386–13396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goujon, C.; Malim, M.H. Characterization of the Alpha Interferon-Induced Postentry Block to HIV-1 Infection in Primary Human Macrophages and T Cells. J. Virol. 2010, 84, 9254–9266. [Google Scholar] [CrossRef] [Green Version]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein Kinase R Reveals an Evolutionary Model for Defeating Viral Mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef]
- Roy, S.; Katze, M.G.; Parkin, N.T.; Edery, I.; Hovanessian, A.G.; Sonenberg, N. Control of the Interferon-Induced 68-Kilodalton Protein Kinase by the HIV-1 Tat Gene Product. Science 1990, 247, 1216–1219. [Google Scholar] [CrossRef]
- Cachat, A.; Alais, S.; Chevalier, S.A.; Journo, C.; Fusil, F.; Dutartre, H.; Boniface, A.; Ko, N.L.; Gessain, A.; Cosset, F.-L.; et al. ADAR1 Enhances HTLV-1 and HTLV-2 Replication through Inhibition of PKR Activity. Retrovirology 2014, 11, 93. [Google Scholar] [CrossRef] [Green Version]
- Valeri, V.W.; Hryniewicz, A.; Andresen, V.; Jones, K.; Fenizia, C.; Bialuk, I.; Chung, H.K.; Fukumoto, R.; Parks, R.W.; Ferrari, M.G.; et al. Requirement of the Human T-Cell Leukemia Virus P12 and P30 Products for Infectivity of Human Dendritic Cells and Macaques but Not Rabbits. Blood 2010, 116, 3809–3817. [Google Scholar] [CrossRef]
- Fenizia, C.; Fiocchi, M.; Jones, K.; Parks, R.W.; Chevalier, S.A.; Ruscetti, F.; Pise-Masison, C.; Franchini, G. Role of HTLV-1 P30 during Infection of Monocytes and Dendritic Cells. Retrovirology 2014, 11, P124. [Google Scholar] [CrossRef] [Green Version]
- Fenizia, C.; Fiocchi, M.; Jones, K.; Parks, R.W.; Ceribelli, M.; Chevalier, S.A.; Edwards, D.; Ruscetti, F.; Pise-Masison, C.A.; Franchini, G. Human T-Cell Leukemia/Lymphoma Virus Type 1 P30, but Not P12/P8, Counteracts Toll-Like Receptor 3 (TLR3) and TLR4 Signaling in Human Monocytes and Dendritic Cells. J. Virol. 2014, 88, 393–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, J.L.; Maldonado, J.O.; Mueller, J.D.; Zhang, W.; Mansky, L.M. Molecular Studies of HTLV-1 Replication: An Update. Viruses 2016, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Wattel, E.; Vartanian, J.P.; Pannetier, C.; Wain-Hobson, S. Clonal Expansion of Human T-Cell Leukemia Virus Type I-Infected Cells in Asymptomatic and Symptomatic Carriers without Malignancy. J. Virol. 1995, 69, 2863–2868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibon, D. HTLV-1 Propels Untransformed CD4+ Lymphocytes into the Cell Cycle While Protecting CD8+ Cells from Death. J. Clin. Investig. 2006, 116, 974–983. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Barez, P.-Y.; Hamaidia, M.; Gazon, H.; de Brogniez, A.; Perike, S.; Gillet, N.; Willems, L. Modes of Human T Cell Leukemia Virus Type 1 Transmission, Replication and Persistence. Viruses 2015, 7, 3603–3624. [Google Scholar] [CrossRef] [Green Version]
- Satou, Y.; Yasunaga, J.; Yoshida, M.; Matsuoka, M. HTLV-I Basic Leucine Zipper Factor Gene MRNA Supports Proliferation of Adult T Cell Leukemia Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Arnold, J.; Zimmerman, B.; Li, M.; Lairmore, M.D.; Green, P.L. Human T-Cell Leukemia Virus Type-1 Antisense-Encoded Gene, Hbz, Promotes T-Lymphocyte Proliferation. Blood 2008, 112, 3788–3797. [Google Scholar] [CrossRef] [Green Version]
- Kuhlmann, A.-S.; Villaudy, J.; Gazzolo, L.; Castellazzi, M.; Mesnard, J.-M.; Duc Dodon, M. HTLV-1 HBZ Cooperates with JunD to Enhance Transcription of the Human Telomerase Reverse Transcriptase Gene (HTERT). Retrovirology 2007, 4, 92. [Google Scholar] [CrossRef] [Green Version]
- Bellon, M.; Yuan, Y.; Nicot, C. Transcription Independent Stimulation of Telomerase Enzymatic Activity by HTLV-I Tax Through Stimulation of IKK. J. Cancer Sci. 2021, 8, 24. [Google Scholar] [CrossRef]
- Yamagishi, M.; Fujikawa, D.; Watanabe, T.; Uchimaru, K. HTLV-1-Mediated Epigenetic Pathway to Adult T-Cell Leukemia–Lymphoma. Front. Microbiol. 2018, 9, 1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamagishi, M.; Suzuki, Y.; Watanabe, T.; Uchimaru, K. Clonal Selection and Evolution of HTLV-1-Infected Cells Driven by Genetic and Epigenetic Alteration. Viruses 2022, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Nozuma, S.; Matsuura, E.; Kodama, D.; Tashiro, Y.; Matsuzaki, T.; Kubota, R.; Izumo, S.; Takashima, H. Effects of Host Restriction Factors and the HTLV-1 Subtype on Susceptibility to HTLV-1-Associated Myelopathy/Tropical Spastic Paraparesis. Retrovirology 2017, 14, 26. [Google Scholar] [CrossRef] [PubMed]
- Leal, F.E.; Menezes, S.M.; Costa, E.A.S.; Brailey, P.M.; Gama, L.; Segurado, A.C.; Kallas, E.G.; Nixon, D.F.; Dierckx, T.; Khouri, R.; et al. Comprehensive Antiretroviral Restriction Factor Profiling Reveals the Evolutionary Imprint of the Ex Vivo and In Vivo IFN-β Response in HTLV-1-Associated Neuroinflammation. Front. Microbiol. 2018, 9, 985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dassouki, Z.; Sahin, U.; El Hajj, H.; Jollivet, F.; Kfoury, Y.; Lallemand-Breitenbach, V.; Hermine, O.; de Thé, H.; Bazarbachi, A. ATL Response to Arsenic/Interferon Therapy Is Triggered by SUMO/PML/RNF4-Dependent Tax Degradation. Blood 2015, 125, 474–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kataoka, K.; Nagata, Y.; Kitanaka, A.; Shiraishi, Y.; Shimamura, T.; Yasunaga, J.-I.; Totoki, Y.; Chiba, K.; Sato-Otsubo, A.; Nagae, G.; et al. Integrated Molecular Analysis of Adult T Cell Leukemia/Lymphoma. Nat. Genet. 2015, 47, 1304–1315. [Google Scholar] [CrossRef]
- Yao, J.; Tanaka, M.; Takenouchi, N.; Ren, Y.; Lee, S.-I.; Fujisawa, J.-I. Induction of APOBEC3B Cytidine Deaminase in HTLV-1-Infected Humanized Mice. Exp. Med. 2019, 17, 3701–3708. [Google Scholar] [CrossRef] [Green Version]
- Mahieux, R.; Suspène, R.; Delebecque, F.; Henry, M.; Schwartz, O.; Wain-Hobson, S.; Vartanian, J.-P. 2005 Extensive Editing of a Small Fraction of Human T-Cell Leukemia Virus Type 1 Genomes by Four APOBEC3 Cytidine Deaminases. J. Gen. Virol. 2005, 86, 2489–2494. [Google Scholar] [CrossRef]
- Fan, J.; Ma, G.; Nosaka, K.; Tanabe, J.; Satou, Y.; Koito, A.; Wain-Hobson, S.; Vartanian, J.-P.; Matsuoka, M. APOBEC3G Generates Nonsense Mutations in Human T-Cell Leukemia Virus Type 1 Proviral Genomes In Vivo. J. Virol. 2010, 84, 7278–7287. [Google Scholar] [CrossRef] [Green Version]
- Poulain, F.; Lejeune, N.; Willemart, K.; Gillet, N.A. Footprint of the Host Restriction Factors APOBEC3 on the Genome of Human Viruses. PLoS Pathog. 2020, 16, e1008718. [Google Scholar] [CrossRef]
- Navarro, F.; Bollman, B.; Chen, H.; König, R.; Yu, Q.; Chiles, K.; Landau, N.R. Complementary Function of the Two Catalytic Domains of APOBEC3G. Virology 2005, 333, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derse, D.; Hill, S.A.; Princler, G.; Lloyd, P.; Heidecker, G. Resistance of Human T Cell Leukemia Virus Type 1 to APOBEC3G Restriction Is Mediated by Elements in Nucleocapsid. Proc. Natl. Acad. Sci. USA 2007, 104, 2915–2920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooms, M.; Krikoni, A.; Kress, A.K.; Simon, V.; Münk, C. APOBEC3A, APOBEC3B, and APOBEC3H Haplotype 2 Restrict Human T-Lymphotropic Virus Type 1. J. Virol. 2012, 86, 6097–6108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gramberg, T.; Kahle, T.; Bloch, N.; Wittmann, S.; Müllers, E.; Daddacha, W.; Hofmann, H.; Kim, B.; Lindemann, D.; Landau, N.R. Restriction of Diverse Retroviruses by SAMHD1. Retrovirology 2013, 10, 26. [Google Scholar] [CrossRef] [Green Version]
- Sze, A.; Belgnaoui, S.M.; Olagnier, D.; Lin, R.; Hiscott, J.; van Grevenynghe, J. Host Restriction Factor SAMHD1 Limits Human T Cell Leukemia Virus Type 1 Infection of Monocytes via STING-Mediated Apoptosis. Cell Host Microbe 2013, 14, 422–434. [Google Scholar] [CrossRef] [Green Version]
- Forlani, G.; Abdallah, R.; Accolla, R.S.; Tosi, G. The MHC-II Transactivator CIITA, a Restriction Factor against Oncogenic HTLV-1 and HTLV-2 Retroviruses: Similarities and Differences in the Inhibition of Tax-1 and Tax-2 Viral Transactivators. Front. Microbiol 2013, 4, 234. [Google Scholar] [CrossRef] [Green Version]
- Forlani, G.; Abdallah, R.; Accolla, R.S.; Tosi, G. The Major Histocompatibility Complex Class II Transactivator CIITA Inhibits the Persistent Activation of NF-ΚB by the Human T Cell Lymphotropic Virus Type 1 Tax-1 Oncoprotein. J. Virol. 2016, 90, 3708–3721. [Google Scholar] [CrossRef] [Green Version]
- Miyazato, P.; Matsuo, M.; Tan, B.J.Y.; Tokunaga, M.; Katsuya, H.; Islam, S.; Ito, J.; Murakawa, Y.; Satou, Y. HTLV-1 Contains a High CG Dinucleotide Content and Is Susceptible to the Host Antiviral Protein ZAP. Retrovirology 2019, 16, 38. [Google Scholar] [CrossRef]
- Ilinskaya, A.; Derse, D.; Hill, S.; Princler, G.; Heidecker, G. Cell-Cell Transmission Allows Human T-Lymphotropic Virus 1 to Circumvent Tetherin Restriction. Virology 2013, 436, 201–209. [Google Scholar] [CrossRef] [Green Version]
- van Gent, M.; Sparrer, K.M.J.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Shi, J.; Friedman, D.B.; Aiken, C. Retrovirus Restriction by TRIM5 Proteins Requires Recognition of Only a Small Fraction of Viral Capsid Subunits. J. Virol. 2013, 87, 9271–9278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skorupka, K.A.; Roganowicz, M.D.; Christensen, D.E.; Wan, Y.; Pornillos, O.; Ganser-Pornillos, B.K. Hierarchical Assembly Governs TRIM5α Recognition of HIV-1 and Retroviral Capsids. Sci. Adv. 2019, 5, 3631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, S.; Kömürlü, S.; Mattick, J.; Selyutina, A.; Talley, S.; Eddins, A.; Diaz-Griffero, F.; Campbell, E.M. K63-Linked Ubiquitin Is Required for Restriction of HIV-1 Reverse Transcription and Capsid Destabilization by Rhesus TRIM5α. J. Virol. 2019, 93, e00558-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A Virus-Packageable CRISPR Screen Identifies Host Factors Mediating Interferon Inhibition of HIV. eLife 2018, 7, e39823. [Google Scholar] [CrossRef] [PubMed]
- Pagani, I.; Poli, G.; Vicenzi, E. TRIM22. A Multitasking Antiviral Factor. Cells 2021, 10, 1864. [Google Scholar] [CrossRef]
- Lim, K.-H.; Park, E.-S.; Kim, D.H.; Cho, K.C.; Kim, K.P.; Park, Y.K.; Ahn, S.H.; Park, S.H.; Kim, K.-H.; Kim, C.W.; et al. Suppression of Interferon-Mediated Anti-HBV Response by Single CpG Methylation in the 5′-UTR of TRIM22. Gut 2018, 67, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.S.; Dudley, J.P. APOBECs and Virus Restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [Green Version]
- Iwatani, Y.; Chan, D.S.B.; Wang, F.; Maynard, K.S.; Sugiura, W.; Gronenborn, A.M.; Rouzina, I.; Williams, M.C.; Musier-Forsyth, K.; Levin, J.G. Deaminase-Independent Inhibition of HIV-1 Reverse Transcription by APOBEC3G. Nucleic Acids Res. 2007, 35, 7096–7108. [Google Scholar] [CrossRef]
- Pollpeter, D.; Parsons, M.; Sobala, A.E.; Coxhead, S.; Lang, R.D.; Bruns, A.M.; Papaioannou, S.; McDonnell, J.M.; Apolonia, L.; Chowdhury, J.A.; et al. Deep Sequencing of HIV-1 Reverse Transcripts Reveals the Multifaceted Anti-Viral Functions of APOBEC3G. Nat. Microbiol. 2018, 3, 220–233. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Choi, J.D.; Malim, M.H. Isolation of a Human Gene That Inhibits HIV-1 Infection and Is Suppressed by the Viral Vif Protein. Nature 2002, 418, 646–650. [Google Scholar] [CrossRef]
- Harris, R.S.; Bishop, K.N.; Sheehy, A.M.; Craig, H.M.; Petersen-Mahrt, S.K.; Watt, I.N.; Neuberger, M.S.; Malim, M.H. DNA Deamination Mediates Innate Immunity to Retroviral Infection. Cell 2003, 113, 803–809. [Google Scholar] [CrossRef] [Green Version]
- Baig, T.T.; Feng, Y.; Chelico, L. Determinants of Efficient Degradation of APOBEC3 Restriction Factors by HIV-1 Vif. J. Virol. 2014, 88, 14380–14395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohsugi, T.; Koito, A. Human T Cell Leukemia Virus Type I Is Resistant to the Antiviral Effects of APOBEC3. J. Virol. Methods 2007, 139, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Ségéral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. SAMHD1 Is the Dendritic- and Myeloid-Cell-Specific HIV-1 Restriction Factor Counteracted by Vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef]
- de Castro-Amarante, M.F.; Pise-Masison, C.A.; McKinnon, K.; Washington Parks, R.; Galli, V.; Omsland, M.; Andresen, V.; Massoud, R.; Brunetto, G.; Caruso, B.; et al. Human T Cell Leukemia Virus Type 1 Infection of the Three Monocyte Subsets Contributes to Viral Burden in Humans. J. Virol. 2015, 90, 2195–2207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuta, R.; Yasunaga, J.; Miura, M.; Sugata, K.; Saito, A.; Akari, H.; Ueno, T.; Takenouchi, N.; Fujisawa, J.; Koh, K.-R.; et al. Human T-Cell Leukemia Virus Type 1 Infects Multiple Lineage Hematopoietic Cells in Vivo. PLoS Pathog. 2017, 13, e1006722. [Google Scholar] [CrossRef] [PubMed]
- Jaguva Vasudevan, A.A.; Becker, D.; Luedde, T.; Gohlke, H.; Münk, C. Foamy Viruses, Bet, and APOBEC3 Restriction. Viruses 2021, 13, 504. [Google Scholar] [CrossRef]
- Bähr, A.; Singer, A.; Hain, A.; Vasudevan, A.A.J.; Schilling, M.; Reh, J.; Riess, M.; Panitz, S.; Serrano, V.; Schweizer, M.; et al. Interferon but Not MxB Inhibits Foamy Retroviruses. Virology 2016, 488, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Delelis, O.; Saïb, A.; Sonigo, P. Biphasic DNA Synthesis in Spumaviruses. J. Virol. 2003, 77, 8141–8146. [Google Scholar] [CrossRef] [Green Version]
- Lori, F.; di Marzo Veronese, F.; de Vico, A.L.; Lusso, P.; Reitz, M.S.; Gallo, R.C. Viral DNA Carried by Human Immunodeficiency Virus Type 1 Virions. J. Virol. 1992, 66, 5067–5074. [Google Scholar] [CrossRef] [Green Version]
- Trono, D. Partial Reverse Transcripts in Virions from Human Immunodeficiency and Murine Leukemia Viruses. J. Virol. 1992, 66, 4893–4900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, E.J.; Mak, J.; Kleiman, L.; Wainberg, M.A. DNA Found in Human Immunodeficiency Virus Type 1 Particles May Not Be Required for Infectivity. J. Gen. Virol. 1994, 75 Pt 7, 1605–1613. [Google Scholar] [CrossRef]
- Pai, R.K.; Askew, D.; Boom, W.H.; Harding, C.V. Regulation of Class II MHC Expression in APCs: Roles of Types I, III, and IV Class II Transactivator. J. Immunol. 2002, 169, 1326–1333. [Google Scholar] [CrossRef] [Green Version]
- Accolla, R.S.; Mazza, S.; De Lerma Barbaro, A.; De Maria, A.; Tosi, G. The HLA Class II Transcriptional Activator Blocks the Function of HIV-1 Tat and Inhibits Viral Replication. Eur. J. Immunol. 2002, 32, 2783–2791. [Google Scholar] [CrossRef]
- Forlani, G.; Turrini, F.; Ghezzi, S.; Tedeschi, A.; Poli, G.; Accolla, R.S.; Tosi, G. The MHC-II Transactivator CIITA Inhibits Tat Function and HIV-1 Replication in Human Myeloid Cells. J. Transl. Med. 2016, 14, 94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlani, G.; Tosi, G.; Turrini, F.; Poli, G.; Vicenzi, E.; Accolla, R.S. Tripartite Motif-Containing Protein 22 Interacts with Class II Transactivator and Orchestrates Its Recruitment in Nuclear Bodies Containing TRIM19/PML and Cyclin T1. Front. Immunol. 2017, 8, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casoli, C.; De Lerma Barbaro, A.; Pilotti, E.; Bertazzoni, U.; Tosi, G.; Accolla, R.S. The MHC Class II Transcriptional Activator (CIITA) Inhibits HTLV-2 Viral Replication by Blocking the Function of the Viral Transactivator Tax-2. Blood 2004, 103, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.; Pilotti, E.; Mortara, L.; Barbaro, A.D.L.; Casoli, C.; Accolla, R.S. Inhibition of Human T Cell Leukemia Virus Type 2 Replication by the Suppressive Action of Class II Transactivator and Nuclear Factor Y. Proc. Natl. Acad. Sci. USA 2006, 103, 12861–12866. [Google Scholar] [CrossRef] [Green Version]
- Tosi, G.; Forlani, G.; Andresen, V.; Turci, M.; Bertazzoni, U.; Franchini, G.; Poli, G.; Accolla, R.S. Major Histocompatibility Complex Class II Transactivator CIITA Is a Viral Restriction Factor That Targets Human T-Cell Lymphotropic Virus Type 1 Tax-1 Function and Inhibits Viral Replication. J. Virol. 2011, 85, 10719–10729. [Google Scholar] [CrossRef] [Green Version]
- Gao, G.; Guo, X.; Goff, S.P. Inhibition of Retroviral RNA Production by ZAP, a CCCH-Type Zinc Finger Protein. Science 2002, 297, 1703–1706. [Google Scholar] [CrossRef]
- Mao, R.; Nie, H.; Cai, D.; Zhang, J.; Liu, H.; Yan, R.; Cuconati, A.; Block, T.M.; Guo, J.-T.; Guo, H. Inhibition of Hepatitis B Virus Replication by the Host Zinc Finger Antiviral Protein. PLOS Pathog. 2013, 9, e1003494. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Ma, X.; Cui, X.; Zhou, J.; Li, C.; Huang, L.; Shang, Y.; Cheng, Z. Inhibition of Avian Tumor Virus Replication by CCCH-Type Zinc Finger Antiviral Protein. Oncotarget 2017, 8, 58865–58871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, M.A.; Gonçalves-Carneiro, D.; Zang, T.M.; Soll, S.J.; York, A.; Blanco-Melo, D.; Bieniasz, P.D. CG Dinucleotide Suppression Enables Antiviral Defence Targeting Non-Self RNA. Nature 2017, 550, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin Inhibits Retrovirus Release and Is Antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klimkait, T.; Strebel, K.; Hoggan, M.D.; Martin, M.A.; Orenstein, J.M. The Human Immunodeficiency Virus Type 1-Specific Protein Vpu Is Required for Efficient Virus Maturation and Release. J. Virol. 1990, 64, 621–629. [Google Scholar] [CrossRef] [Green Version]
- Takeda, E.; Nakagawa, S.; Nakaya, Y.; Tanaka, A.; Miyazawa, T.; Yasuda, J. Identification and Functional Analysis of Three Isoforms of Bovine BST-2. PLoS ONE 2012, 7, e41483. [Google Scholar] [CrossRef] [Green Version]
- Liang, Z.; Zhang, Y.; Song, J.; Zhang, H.; Zhang, S.; Li, Y.; Tan, J.; Qiao, W. The Effect of Bovine BST2A1 on the Release and Cell-to-Cell Transmission of Retroviruses. Virol. J. 2017, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Maali, Y.; Journo, C.; Mahieux, R.; Dutartre, H. Microbial Biofilms: Human T-Cell Leukemia Virus Type 1 First in Line for Viral Biofilm but Far Behind Bacterial Biofilms. Front. Microbiol. 2020, 11, 2041. [Google Scholar] [CrossRef]
- van Montfoort, N.; Olagnier, D.; Hiscott, J. Unmasking Immune Sensing of Retroviruses: Interplay between Innate Sensors and Host Effectors. Cytokine Growth Factor Rev. 2014, 25, 657–668. [Google Scholar] [CrossRef]
- Colisson, R.; Barblu, L.; Gras, C.; Raynaud, F.; Hadj-Slimane, R.; Pique, C.; Hermine, O.; Lepelletier, Y.; Herbeuval, J.-P. Free HTLV-1 Induces TLR7-Dependent Innate Immune Response and TRAIL Relocalization in Killer Plasmacytoid Dendritic Cells. Blood 2010, 115, 2177–2185. [Google Scholar] [CrossRef] [Green Version]
- Assil, S.; Futsch, N.; Décembre, E.; Alais, S.; Gessain, A.; Cosset, F.-L.; Mahieux, R.; Dreux, M.; Dutartre, H. Sensing of Cell-Associated HTLV by Plasmacytoid Dendritic Cells Is Regulated by Dense β-Galactoside Glycosylation. PLOS Pathog. 2019, 15, e1007589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Song, D.; Liu, Y.; Cui, Y.; Lu, G.; Di, W.; Xing, H.; Ma, L.; Guo, Z.; Guan, Y.; et al. IFI16 Regulates HTLV-1 Replication through Promoting HTLV-1 RTI-Induced Innate Immune Responses. FEBS Lett. 2018, 592, 1693–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Yang, S.; Liu, L.; Wang, H.; Yang, B. HTLV-1 Tax Impairs K63-Linked Ubiquitination of STING to Evade Host Innate Immunity. Virus Res. 2017, 232, 13–21. [Google Scholar] [CrossRef]
- Banchereau, R.; Baldwin, N.; Cepika, A.-M.; Athale, S.; Xue, Y.; Yu, C.I.; Metang, P.; Cheruku, A.; Berthier, I.; Gayet, I.; et al. Transcriptional Specialization of Human Dendritic Cell Subsets in Response to Microbial Vaccines. Nat. Commun. 2014, 5, 5283. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.-L.; Liang, C. The IFITM Proteins Inhibit HIV-1 Infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [Green Version]
- Goujon, C.; Moncorgé, O.; Bauby, H.; Doyle, T.; Ward, C.C.; Schaller, T.; Hué, S.; Barclay, W.S.; Schulz, R.; Malim, M.H. Human MX2 Is an Interferon-Induced Post-Entry Inhibitor of HIV-1 Infection. Nature 2013, 502, 559–562. [Google Scholar] [CrossRef]
- Kane, M.; Yadav, S.S.; Bitzegeio, J.; Kutluay, S.B.; Zang, T.; Wilson, S.J.; Schoggins, J.W.; Rice, C.M.; Yamashita, M.; Hatziioannou, T.; et al. MX2 Is an Interferon-Induced Inhibitor of HIV-1 Infection. Nature 2013, 502, 563–566. [Google Scholar] [CrossRef] [Green Version]
- Rosa, A.; Chande, A.; Ziglio, S.; De Sanctis, V.; Bertorelli, R.; Goh, S.L.; McCauley, S.M.; Nowosielska, A.; Antonarakis, S.E.; Luban, J.; et al. HIV-1 Nef Promotes Infection by Excluding SERINC5 from Virion Incorporation. Nature 2015, 526, 212–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Usami, Y.; Wu, Y.; Göttlinger, H.G. SERINC3 and SERINC5 Restrict HIV-1 Infectivity and Are Counteracted by Nef. Nature 2015, 526, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.H.; Miller, C.; Bankers, L.; Crameri, G.; Wang, L.-F.; Poeschla, E.M. A Potent Postentry Restriction to Primate Lentiviruses in a Yinpterochiropteran Bat. mBio 2020, 11, e01854-20. [Google Scholar] [CrossRef]
- Dong, L.; Cheng, Q.; Wang, Z.; Yuan, P.; Li, Z.; Sun, Y.; Han, S.; Yin, J.; Peng, B.; He, X.; et al. Human Pirh2 Is A Novel Inhibitor of Prototype Foamy Virus Replication. Viruses 2015, 7, 1668–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, D.; Goff, S.P. Embryonic Stem Cells Use ZFP809 to Silence Retroviral DNAs. Nature 2009, 458, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Yan, J.; Wang, S.; Guo, Y.; Xi, X.; Han, S.; Yin, J.; Peng, B.; He, X.; Bodem, J.; et al. Trim28 Acts as Restriction Factor of Prototype Foamy Virus Replication by Modulating H3K9me3 Marks and Destabilizing the Viral Transactivator Tas. Retrovirology 2021, 18, 38. [Google Scholar] [CrossRef] [PubMed]
- Alais, S.; Pasquier, A.; Jegado, B.; Journo, C.; Rua, R.; Gessain, A.; Tobaly-Tapiero, J.; Lacoste, R.; Turpin, J.; Mahieux, R. STLV-1 Co-Infection Is Correlated with an Increased SFV Proviral Load in the Peripheral Blood of SFV/STLV-1 Naturally Infected Non-Human Primates. PLOS Negl. Trop. Dis. 2018, 12, e0006812. [Google Scholar] [CrossRef]
- Hron, T.; Farkašová, H.; Gifford, R.J.; Benda, P.; Hulva, P.; Görföl, T.; Pačes, J.; Elleder, D. Remnants of an Ancient Deltaretrovirus in the Genomes of Horseshoe Bats (Rhinolophidae). Viruses 2018, 10, 185. [Google Scholar] [CrossRef] [Green Version]
- Hron, T.; Elleder, D.; Gifford, R.J. Deltaretroviruses Have Circulated since at Least the Paleogene and Infected a Broad Range of Mammalian Species. Retrovirology 2019, 16, 33. [Google Scholar] [CrossRef]
- Farkašová, H.; Hron, T.; Pačes, J.; Hulva, P.; Benda, P.; Gifford, R.J.; Elleder, D. Discovery of an Endogenous Deltaretrovirus in the Genome of Long-Fingered Bats (Chiroptera: Miniopteridae). Proc. Natl. Acad. Sci. USA 2017, 114, 3145–3150. [Google Scholar] [CrossRef] [Green Version]
- Coon, S.; Wang, D.; Wu, L. Polymorphisms of the SAMHD1 Gene Are Not Associated with the Infection and Natural Control of HIV Type 1 in Europeans and African-Americans. AIDS Res. Hum. Retrovir. 2012, 28, 1565–1573. [Google Scholar] [CrossRef] [Green Version]
- Sabouri, A.H.; Saito, M.; Lloyd, A.L.; Vine, A.M.; Witkover, A.W.; Furukawa, Y.; Izumo, S.; Arimura, K.; Marshall, S.E.F.; Usuku, K.; et al. Polymorphism in the Interleukin-10 Promoter Affects Both Provirus Load and the Risk of Human T Lymphotropic Virus Type I-Associated Myelopathy/Tropical Spastic Paraparesis. J. Infect. Dis. 2004, 190, 1279–1285. [Google Scholar] [CrossRef]
- Savoy, S.K.A.; Boudreau, J.E. The Evolutionary Arms Race between Virus and NK Cells: Diversity Enables Population-Level Virus Control. Viruses 2019, 11, 959. [Google Scholar] [CrossRef] [Green Version]
- Gallo, R.C. Human Retroviruses after 20 Years: A Perspective from the Past and Prospects for Their Future Control. Immunol. Rev. 2002, 185, 236–265. [Google Scholar] [CrossRef]
- Mansky, L.M. In Vivo Analysis of Human T-Cell Leukemia Virus Type 1 Reverse Transcription Accuracy. J. Virol. 2000, 74, 9525–9531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melamed, A.; Laydon, D.J.; Al Khatib, H.; Rowan, A.G.; Taylor, G.P.; Bangham, C.R. HTLV-1 Drives Vigorous Clonal Expansion of Infected CD8(+) T Cells in Natural Infection. Retrovirology 2015, 12, 91. [Google Scholar] [CrossRef]
- Richardson, J.H.; Höllsberg, P.; Windhagen, A.; Child, L.A.; Hafler, D.A.; Lever, A.M.L. Variable Immortalizing Potential and Frequent Virus Latency in Blood-Derived T-Cell Clones Infected With Human T-Cell Leukemia Virus Type I. Blood 1997, 89, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Gillet, N.A.; Malani, N.; Melamed, A.; Gormley, N.; Carter, R.; Bentley, D.; Berry, C.; Bushman, F.D.; Taylor, G.P.; Bangham, C.R. The Host Genomic Environment of the Provirus Determines the Abundance of HTLV-1-Infected T-Cell Clones. Blood 2011, 117, 3113–3122. [Google Scholar] [CrossRef] [PubMed]
- Melamed, A.; Laydon, D.J.; Gillet, N.A.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R.M. Genome-Wide Determinants of Proviral Targeting, Clonal Abundance and Expression in Natural HTLV-1 Infection. PLOS Pathog. 2013, 9, e1003271. [Google Scholar] [CrossRef] [Green Version]
- Bangham, C.R.M.; Miura, M.; Kulkarni, A.; Matsuoka, M. Regulation of Latency in the Human T Cell Leukemia Virus, HTLV-1. Annu. Rev. Virol. 2019, 6, 365–385. [Google Scholar] [CrossRef]
- Fauquenoy, S.; Robette, G.; Kula, A.; Vanhulle, C.; Bouchat, S.; Delacourt, N.; Rodari, A.; Marban, C.; Schwartz, C.; Burny, A.; et al. Repression of Human T-Lymphotropic Virus Type 1 Long Terminal Repeat Sense Transcription by Sp1 Recruitment to Novel Sp1 Binding Sites. Sci. Rep. 2017, 7, 43221. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Santoso, N.; Power, D.; Simpson, S.; Dieringer, M.; Miao, H.; Gurova, K.; Giam, C.-Z.; Elledge, S.J.; Zhu, J. FACT Proteins, SUPT16H and SSRP1, Are Transcriptional Suppressors of HIV-1 and HTLV-1 That Facilitate Viral Latency. J. Biol. Chem. 2015, 290, 27297–27310. [Google Scholar] [CrossRef] [Green Version]
- Kulkarni, A.; Taylor, G.P.; Klose, R.J.; Schofield, C.J.; Bangham, C.R. Histone H2A Monoubiquitylation and P38-MAPKs Regulate Immediate-Early Gene-like Reactivation of Latent Retrovirus HTLV-1. JCI Insight 2018, 3, e123196. [Google Scholar] [CrossRef]
- Chougui, G.; Margottin-Goguet, F. HUSH, a Link Between Intrinsic Immunity and HIV Latency. Front. Microbiol. 2019, 10, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moles, R.; Sarkis, S.; Galli, V.; Omsland, M.; Artesi, M.; Bissa, M.; McKinnon, K.; Brown, S.; Hahaut, V.; Washington-Parks, R.; et al. NK Cells and Monocytes Modulate Primary HTLV-1 Infection. PLOS Pathog. 2022, 18, e1010416. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Antiviral Factor | Mechanism of the Antiviral Effect on HTLV-1 | References |
|---|---|---|
| TRIM family | TRIM5α may restrict HTLV-1 replication | [43,44] |
| TRIM19/PML restricts HTLV-1 viral expression through Tax degradation | [45] | |
| APOBEC3 | No differences in A3G expression regarding the clinical status or the PVL | [46,47] |
| Hypermutations of viral genomes indicating an evolutionary APOBEC3 editing in PBMC from asymptomatic carriers or ATL patients | [48,49,50] | |
| HTLV-1 is relatively resistant to A3G antiviral activity despite A3G being packaged into HTLV-1 virions | [48,51,52] | |
| A3A, A3B and A3H hapII are packaged into HTLV-1 virions and reduce viral replication in target cells | [53] | |
| SAMHD1 | No restriction by SAMHD1 in macrophages and dendritic cells | [54] |
| Restriction of monocyte infection in vitro via SAMHD1-dependent abortive reverse transcription and induction of apoptosis | [55] | |
| CIITA | CIITA restricts HTLV-1 viral expression through inhibition of Tax transactivating activity | [56,57] |
| ZAP | ZAP restricts HTLV-1 viral production, most probably by processing HTLV-1 transcripts | [58] |
| Tetherin | No restriction by Tetherin; in contrast, it may favor HTLV-1 spread | [59] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carcone, A.; Journo, C.; Dutartre, H. Is the HTLV-1 Retrovirus Targeted by Host Restriction Factors? Viruses 2022, 14, 1611. https://doi.org/10.3390/v14081611
Carcone A, Journo C, Dutartre H. Is the HTLV-1 Retrovirus Targeted by Host Restriction Factors? Viruses. 2022; 14(8):1611. https://doi.org/10.3390/v14081611
Chicago/Turabian StyleCarcone, Auriane, Chloé Journo, and Hélène Dutartre. 2022. "Is the HTLV-1 Retrovirus Targeted by Host Restriction Factors?" Viruses 14, no. 8: 1611. https://doi.org/10.3390/v14081611