
Crocodilepox Virus Protein 157 Is an Independently Evolved Inhibitor of Protein Kinase R


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Plasmids
2.3. Transfection and Luciferase-Based Reporter (LBR) Assays
2.4. Viruses and Infection Assays
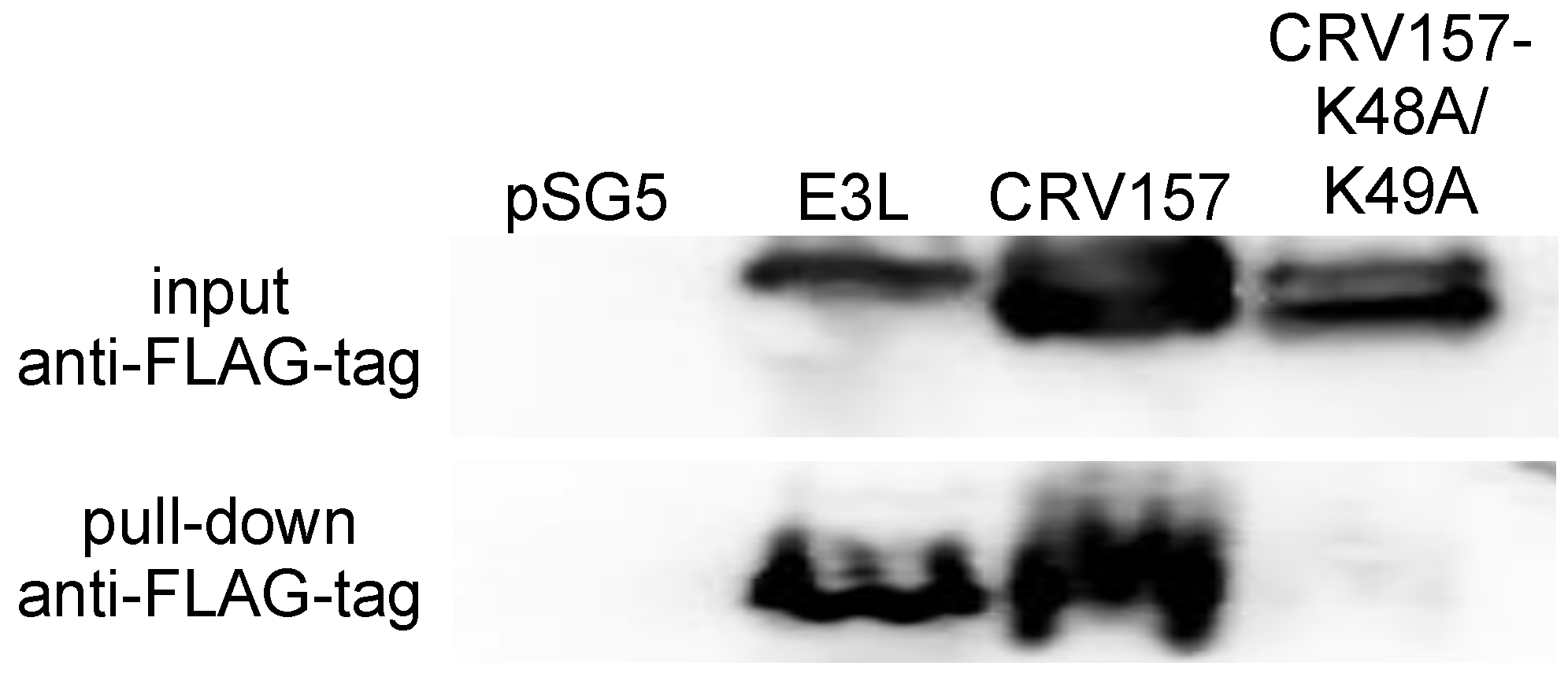
2.5. Poly I:C Pull-Down Assays
2.6. Western Blots
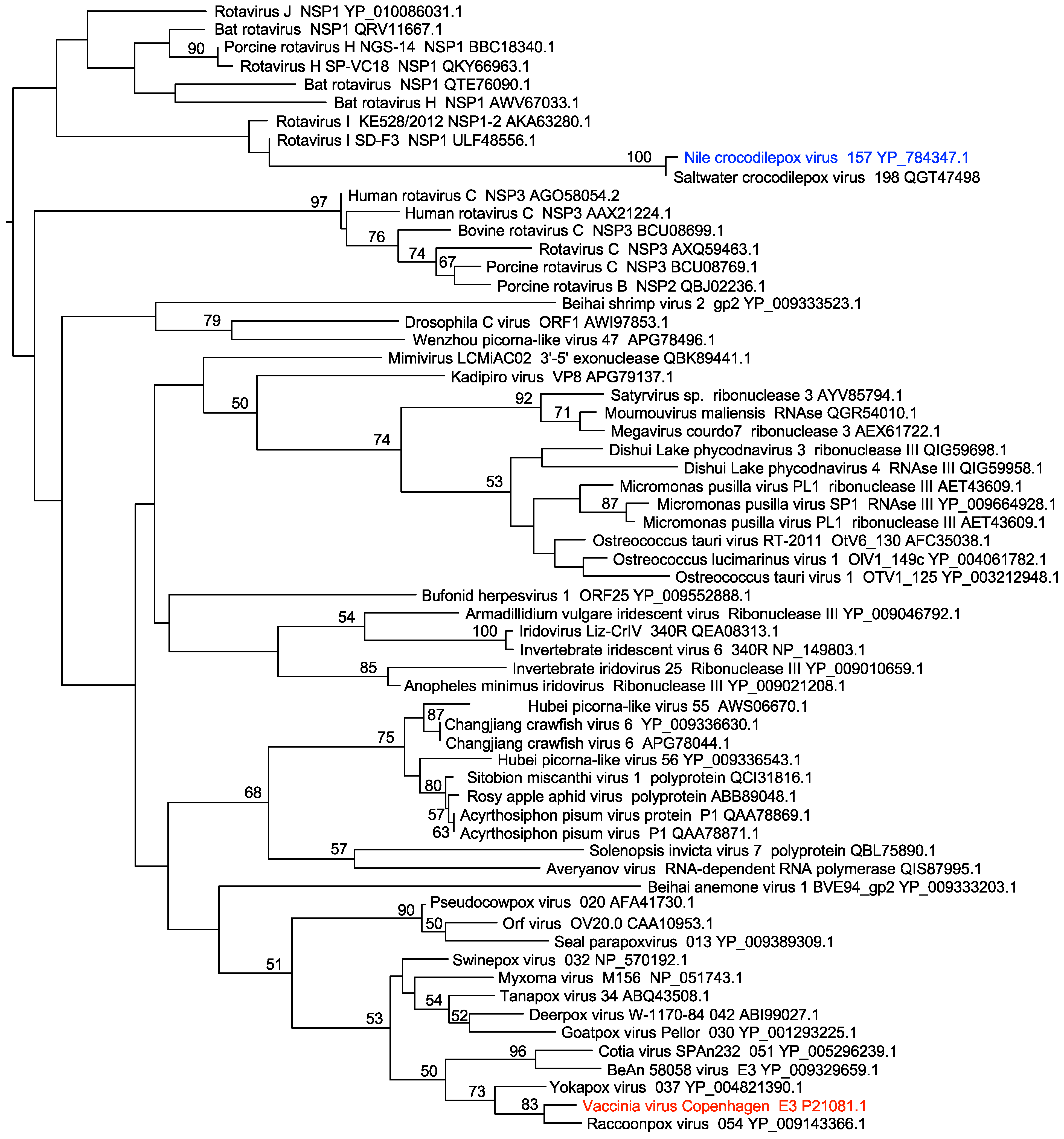
2.7. Phylogenetic Analysis
2.8. Confocal Microscopy
2.9. Quantitative Colocalization Analysis (QCA)
3. Results
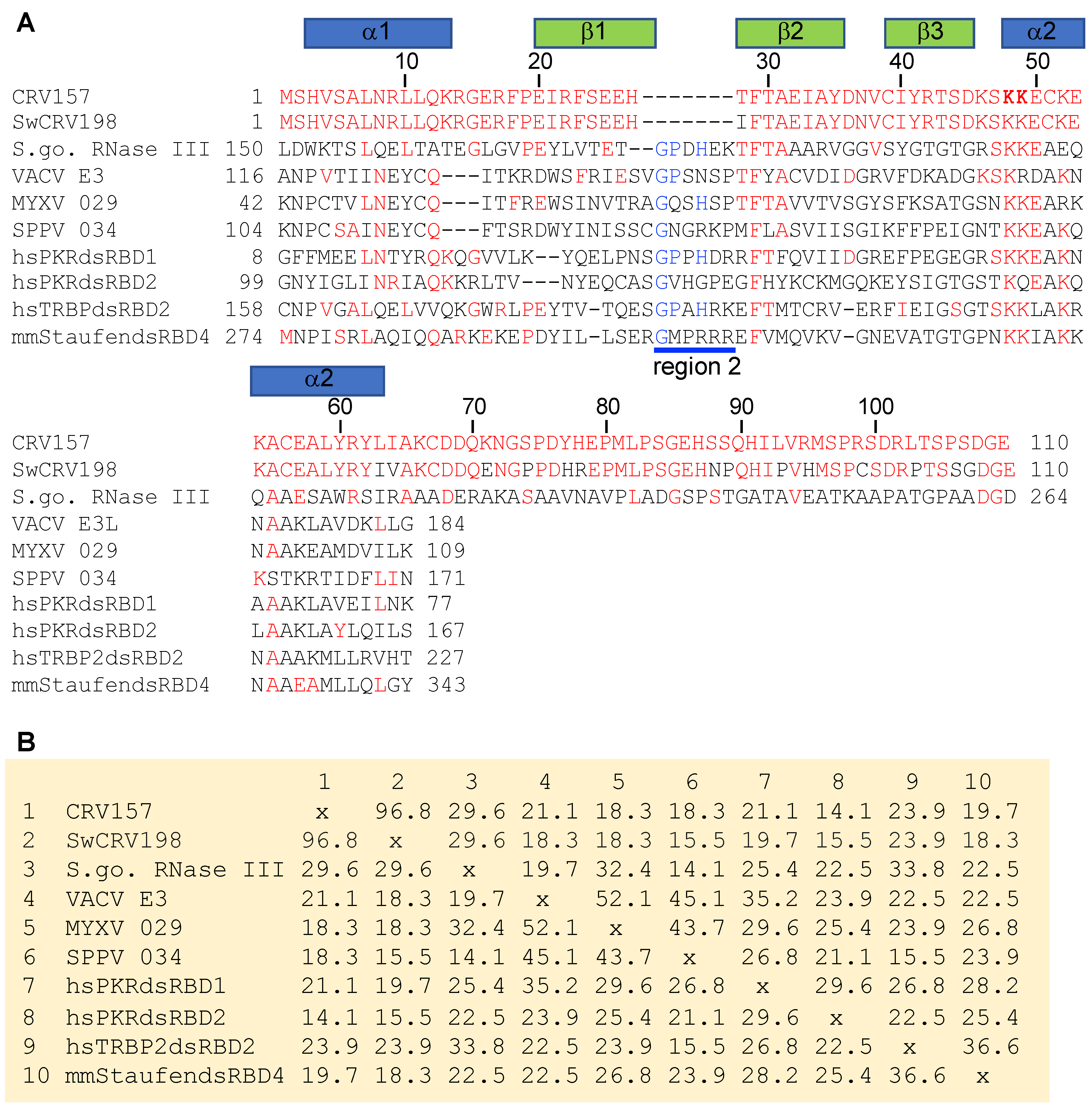
3.1. CRV157 Contains a dsRNA-Binding Domain
3.2. CRV157 Is a dsRNA Binding Protein
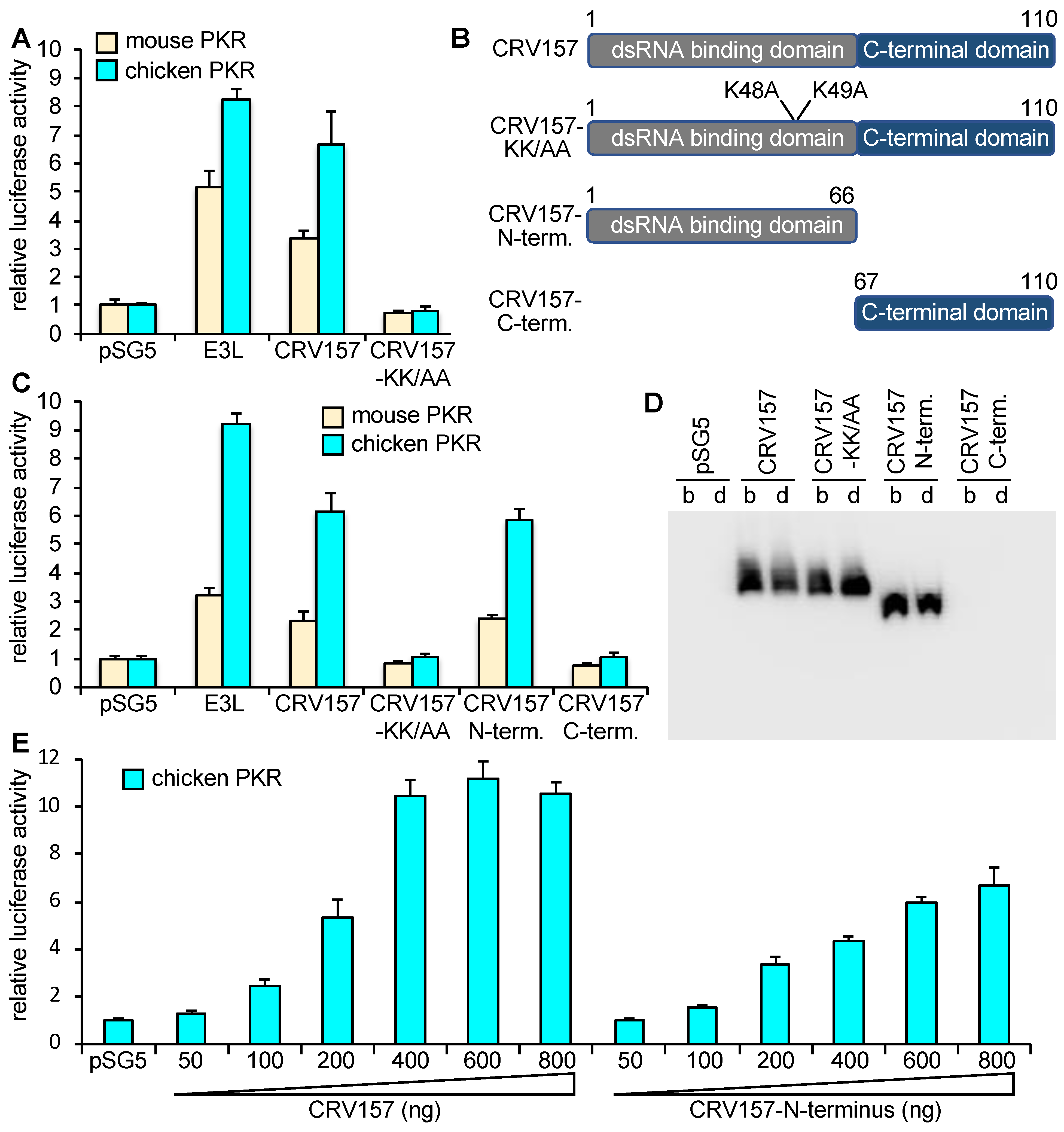
3.3. CRV157 Is a PKR Inhibitor
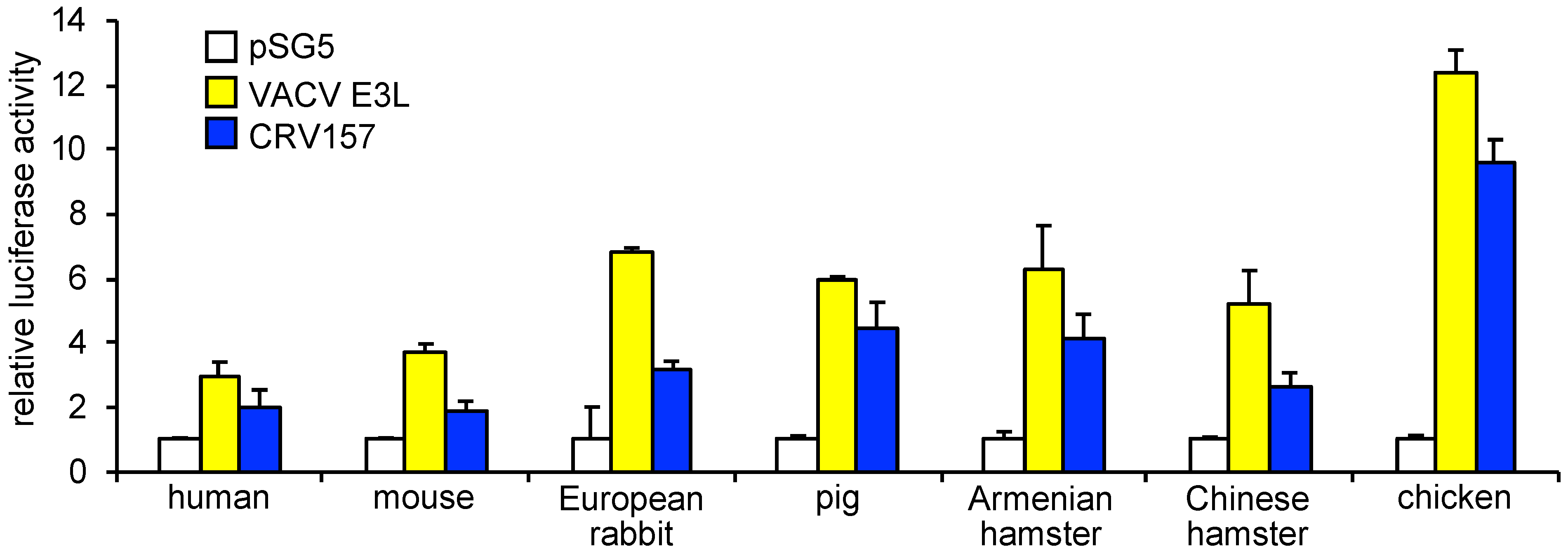
3.4. CRV157 Can Inhibit PKR from Multiple Species
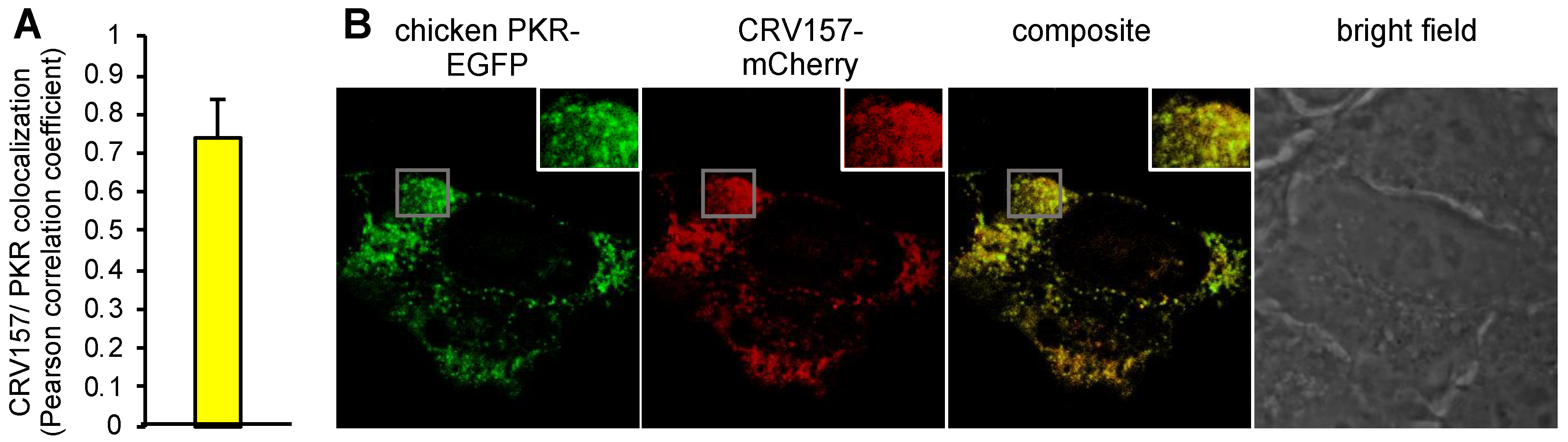
3.5. Colocalization of CRV157 with PKR
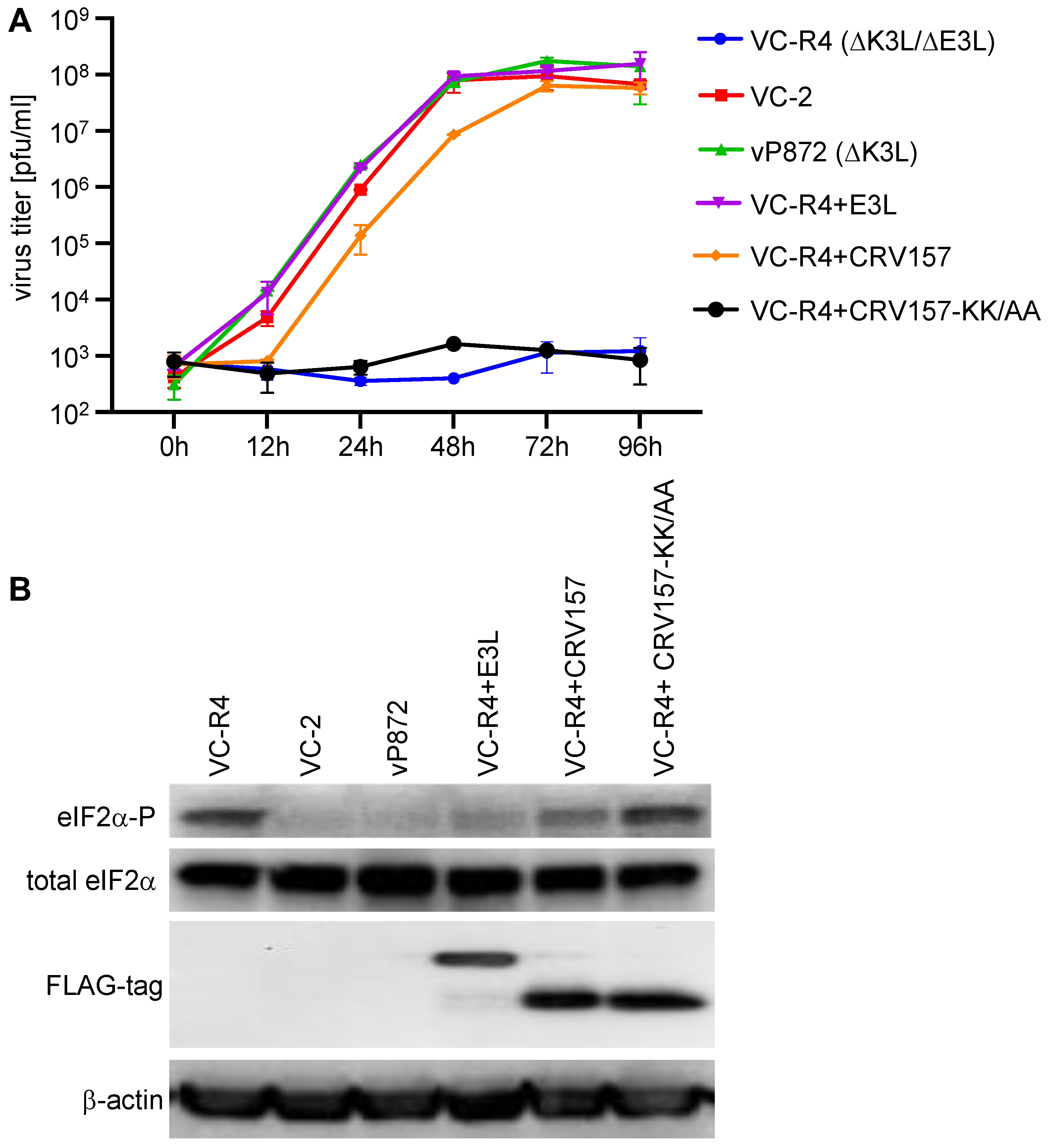
3.6. CRV157 Can Rescue the Replication of an Attenuated VACV Strain That Lacks Its PKR Inhibitors
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hendrickson, R.C.; Wang, C.; Hatcher, E.L.; Lefkowitz, E.J. Orthopoxvirus genome evolution: The role of gene loss. Viruses 2010, 2, 1933–1967. [Google Scholar] [CrossRef]
- McFadden, G. Poxvirus tropism. Nat. Rev. Microbiol. 2005, 3, 201–213. [Google Scholar] [CrossRef]
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef] [Green Version]
- Silva, N.I.O.; de Oliveira, J.S.; Kroon, E.G.; Trindade, G.S.; Drumond, B.P. Here, There, and Everywhere: The Wide Host Range and Geographic Distribution of Zoonotic Orthopoxviruses. Viruses 2020, 13, 43. [Google Scholar] [CrossRef]
- Jacobson, E.R.; Popp, J.A.; Shields, R.P.; Gaskin, J.M. Poxlike skin lesions in captive caimans. J. Am. Vet. Med. Assoc. 1979, 175, 937–940. [Google Scholar]
- Pandey, G.S.; Inoue, N.; Ohshima, K.; Okada, K.; Chihaya, Y.; Fujimoto, Y. Poxvirus infection in Nile crocodiles (Crocodylus niloticus). Res. Vet. Sci. 1990, 49, 171–176. [Google Scholar] [CrossRef]
- Penrith, M.L.; Nesbit, J.W.; Huchzermeyer, F.W. Pox virus infection in captive juvenile caimans (Caiman crocodilus fuscus) in South Africa. J. S. Afr. Vet. Assoc. 1991, 62, 137–139. [Google Scholar] [CrossRef]
- Huchzermeyer, F.W.; Wallace, D.B.; Putterill, J.F.; Gerdes, G.H. Identification and partial sequencing of a crocodile poxvirus associated with deeply penetrating skin lesions in farmed Nile crocodiles, Crocodylus niloticus. Onderstepoort J. Vet. Res. 2009, 76, 311–316. [Google Scholar] [CrossRef] [Green Version]
- Marschang, R.E. Viruses infecting reptiles. Viruses 2011, 3, 2087–2126. [Google Scholar] [CrossRef] [Green Version]
- Afonso, C.L.; Tulman, E.R.; Delhon, G.; Lu, Z.; Viljoen, G.J.; Wallace, D.B.; Kutish, G.F.; Rock, D.L. Genome of crocodilepox virus. J. Virol. 2006, 80, 4978–4991. [Google Scholar] [CrossRef] [Green Version]
- Sarker, S.; Isberg, S.R.; Milic, N.L.; Lock, P.; Helbig, K.J. Molecular characterization of the first saltwater crocodilepox virus genome sequences from the world’s largest living member of the Crocodylia. Sci. Rep. 2018, 8, 5623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huchzermeyer, F.W.; Huchzermeyer, K.D.; Putterill, J.F. Observations on a field outbreak of pox virus infection in young Nile crocodiles (Crocodylus niloticus). J. S. Afr. Vet. Assoc. 1991, 62, 27–29. [Google Scholar] [CrossRef]
- Huchzermeyer, F.W. Diseases of farmed crocodiles and ostriches. Rev. Sci. Tech. 2002, 21, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Horner, R.F. Poxvirus in farmed Nile crocodiles. Vet. Rec. 1988, 122, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, G.H. Morphology of poxviruses from reptiles. Vet. Rec. 1991, 128, 452. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.L.; Isberg, S.R.; Shilton, C.M.; Milic, N.L. Impact of poxvirus lesions on saltwater crocodile (Crocodylus porosus) skins. Vet. Microbiol. 2017, 211, 29–35. [Google Scholar] [CrossRef]
- Moss, B. Poxvirus entry and membrane fusion. Virology 2006, 344, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Bowie, A.G.; Unterholzner, L. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Yu, H.; Bruneau, R.C.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. [Google Scholar] [CrossRef]
- Meurs, E.; Chong, K.; Galabru, J.; Thomas, N.S.; Kerr, I.M.; Williams, B.R.; Hovanessian, A.G. Molecular cloning and characterization of the human double-stranded RNA-activated protein kinase induced by interferon. Cell 1990, 62, 379–390. [Google Scholar] [CrossRef]
- Beattie, E.; Tartaglia, J.; Paoletti, E. Vaccinia virus-encoded eIF-2 alpha homolog abrogates the antiviral effect of interferon. Virology 1991, 183, 419–422. [Google Scholar] [CrossRef]
- Chang, H.W.; Watson, J.C.; Jacobs, B.L. The E3L gene of vaccinia virus encodes an inhibitor of the interferon-induced, double-stranded RNA-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1992, 89, 4825–4829. [Google Scholar] [CrossRef] [Green Version]
- Davies, M.V.; Chang, H.W.; Jacobs, B.L.; Kaufman, R.J. The E3L and K3L vaccinia virus gene products stimulate translation through inhibition of the double-stranded RNA-dependent protein kinase by different mechanisms. J. Virol. 1993, 67, 1688–1692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langland, J.O.; Cameron, J.M.; Heck, M.C.; Jancovich, J.K.; Jacobs, B.L. Inhibition of PKR by RNA and DNA viruses. Virus Res. 2006, 119, 100–110. [Google Scholar] [CrossRef]
- Kerr, I.M.; Brown, R.E.; Hovanessian, A.G. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature 1977, 268, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Roberts, W.K.; Hovanessian, A.; Brown, R.E.; Clemens, M.J.; Kerr, I.M. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature 1976, 264, 477–480. [Google Scholar] [CrossRef]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem. 1987, 262, 15538–15544. [Google Scholar] [CrossRef]
- Wu, S.; Kaufman, R.J. A model for the double-stranded RNA (dsRNA)-dependent dimerization and activation of the dsRNA-activated protein kinase PKR. J. Biol. Chem. 1997, 272, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Dar, A.C.; Dever, T.E.; Sicheri, F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell 2005, 122, 887–900. [Google Scholar] [CrossRef] [Green Version]
- Dey, M.; Cao, C.; Dar, A.C.; Tamura, T.; Ozato, K.; Sicheri, F.; Dever, T.E. Mechanistic link between PKR dimerization, autophosphorylation, and eIF2alpha substrate recognition. Cell 2005, 122, 901–913. [Google Scholar] [CrossRef] [Green Version]
- Garcia, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of protein kinase PKR in cell biology: From antiviral to antiproliferative action. Microbiol. Mol. Biol. Rev. 2006, 70, 1032–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, M.; Mann, B.R.; Anshu, A.; Mannan, M.A. Activation of protein kinase PKR requires dimerization-induced cis-phosphorylation within the activation loop. J. Biol. Chem. 2014, 289, 5747–5757. [Google Scholar] [CrossRef] [Green Version]
- Guerra, S.; Lopez-Fernandez, L.A.; Conde, R.; Pascual-Montano, A.; Harshman, K.; Esteban, M. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J. Virol. 2004, 78, 5820–5834. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.B.; Esteban, M. The interferon-induced double-stranded RNA-activated protein kinase induces apoptosis. Virology 1994, 199, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Samuel, C.E. Protein kinase PKR plays a stimulus- and virus-dependent role in apoptotic death and virus multiplication in human cells. J. Virol. 2007, 81, 8192–8200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Peng, C.; Zhang, C.; Stoian, A.M.M.; Tazi, L.; Brennan, G.; Rothenburg, S. Maladaptation after a virus host switch leads to increased activation of the pro-inflammatory NF-kappaB pathway. Proc. Natl. Acad. Sci. USA 2022, 119, e2115354119. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A.; Rothenburg, S. A survey of host range genes in poxvirus genomes. Infect. Genet. Evol. 2013, 14, 406–425. [Google Scholar] [CrossRef] [Green Version]
- Senkevich, T.G.; Yutin, N.; Wolf, Y.I.; Koonin, E.V.; Moss, B. Ancient Gene Capture and Recent Gene Loss Shape the Evolution of Orthopoxvirus-Host Interaction Genes. mBio 2021, 12, e0149521. [Google Scholar] [CrossRef]
- Watson, J.C.; Chang, H.W.; Jacobs, B.L. Characterization of a vaccinia virus-encoded double-stranded RNA-binding protein that may be involved in inhibition of the double-stranded RNA-dependent protein kinase. Virology 1991, 185, 206–216. [Google Scholar] [CrossRef]
- Chang, H.W.; Jacobs, B.L. Identification of a conserved motif that is necessary for binding of the vaccinia virus E3L gene products to double-stranded RNA. Virology 1993, 194, 537–547. [Google Scholar] [CrossRef]
- Romano, P.R.; Zhang, F.; Tan, S.L.; Garcia-Barrio, M.T.; Katze, M.G.; Dever, T.E.; Hinnebusch, A.G. Inhibition of double-stranded RNA-dependent protein kinase PKR by vaccinia virus E3: Role of complex formation and the E3 N-terminal domain. Mol. Cell Biol. 1998, 18, 7304–7316. [Google Scholar] [CrossRef] [Green Version]
- Dar, A.C.; Sicheri, F. X-ray crystal structure and functional analysis of vaccinia virus K3L reveals molecular determinants for PKR subversion and substrate recognition. Mol. Cell 2002, 10, 295–305. [Google Scholar] [CrossRef]
- Little, N.S.; Quon, T.; Upton, C. Prediction of a novel RNA binding domain in crocodilepox Zimbabwe Gene 157. Microb. Inform. Exp. 2011, 1, 12. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.M.; Liu, J.; Chan, W.M.; Rothenburg, S.; McFadden, G. Myxoma virus protein M029 is a dual function immunomodulator that inhibits PKR and also conscripts RHA/DHX9 to promote expanded host tropism and viral replication. PLoS Pathog. 2013, 9, e1003465. [Google Scholar] [CrossRef] [PubMed]
- Rothenburg, S.; Seo, E.J.; Gibbs, J.S.; Dever, T.E.; Dittmar, K. Rapid evolution of protein kinase PKR alters sensitivity to viral inhibitors. Nat. Struct. Mol. Biol. 2009, 16, 63–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.; Haller, S.L.; Rahman, M.M.; McFadden, G.; Rothenburg, S. Myxoma virus M156 is a specific inhibitor of rabbit PKR but contains a loss-of-function mutation in Australian virus isolates. Proc. Natl. Acad. Sci. USA 2016, 113, 3855–3860. [Google Scholar] [CrossRef] [Green Version]
- Vipat, S.; Brennan, G.; Park, C.; Haller, S.L.; Rothenburg, S. Rapid, Seamless Generation of Recombinant Poxviruses using Host Range and Visual Selection. J. Vis. Exp. 2020, 24, e61049. [Google Scholar] [CrossRef]
- Dueck, K.J.; Hu, Y.S.; Chen, P.; Deschambault, Y.; Lee, J.; Varga, J.; Cao, J. Mutational analysis of vaccinia virus E3 protein: The biological functions do not correlate with its biochemical capacity to bind double-stranded RNA. J. Virol. 2015, 89, 5382–5394. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolte, S.; Cordelieres, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Tian, B.; Mathews, M.B. Phylogenetics and functions of the double-stranded RNA-binding motif: A genomic survey. Prog Nucleic Acid Res. Mol. Biol. 2003, 74, 123–158. [Google Scholar] [CrossRef] [PubMed]
- Masliah, G.; Barraud, P.; Allain, F.H. RNA recognition by double-stranded RNA binding domains: A matter of shape and sequence. Cell Mol. Life Sci. 2013, 70, 1875–1895. [Google Scholar] [CrossRef] [Green Version]
- Bycroft, M.; Grunert, S.; Murzin, A.G.; Proctor, M.; St Johnston, D. NMR solution structure of a dsRNA binding domain from Drosophila staufen protein reveals homology to the N-terminal domain of ribosomal protein S5. EMBO J. 1995, 14, 3563–3571. [Google Scholar] [CrossRef]
- Ho, C.K.; Shuman, S. Mutational analysis of the vaccinia virus E3 protein defines amino acid residues involved in E3 binding to double-stranded RNA. J. Virol. 1996, 70, 2611–2614. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.; Grunert, S.; Adams, J.; Micklem, D.R.; Proctor, M.R.; Freund, S.; Bycroft, M.; St Johnston, D.; Varani, G. RNA recognition by a Staufen double-stranded RNA-binding domain. EMBO J. 2000, 19, 997–1009. [Google Scholar] [CrossRef] [Green Version]
- Nejepinska, J.; Malik, R.; Wagner, S.; Svoboda, P. Reporters transiently transfected into mammalian cells are highly sensitive to translational repression induced by dsRNA expression. PLoS ONE 2014, 9, e87517. [Google Scholar] [CrossRef]
- Elde, N.C.; Child, S.J.; Geballe, A.P.; Malik, H.S. Protein kinase R reveals an evolutionary model for defeating viral mimicry. Nature 2009, 457, 485–489. [Google Scholar] [CrossRef]
- Park, C.; Peng, C.; Brennan, G.; Rothenburg, S. Species-specific inhibition of antiviral protein kinase R by capripoxviruses and vaccinia virus. Ann. N. Y. Acad. Sci. 2019, 1438, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Park, C.; Peng, C.; Rahman, M.J.; Haller, S.L.; Tazi, L.; Brennan, G.; Rothenburg, S. Orthopoxvirus K3 orthologs show virus- and host-specific inhibition of the antiviral protein kinase PKR. PLoS Pathog. 2021, 17, e1009183. [Google Scholar] [CrossRef] [PubMed]
- Mukaka, M.M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Sarker, S.; Isberg, S.R.; Moran, J.L.; Araujo, R.; Elliott, N.; Melville, L.; Beddoe, T.; Helbig, K.J. Crocodilepox Virus Evolutionary Genomics Supports Observed Poxvirus Infection Dynamics on Saltwater Crocodile (Crocodylus porosus). Viruses 2019, 11, 1116. [Google Scholar] [CrossRef] [Green Version]
- Odom, M.R.; Hendrickson, R.C.; Lefkowitz, E.J. Poxvirus protein evolution: Family wide assessment of possible horizontal gene transfer events. Virus Res. 2009, 144, 233–249. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Koonin, E.V.; Moss, B. Vaccinia virus F16 protein, a predicted catalytically inactive member of the prokaryotic serine recombinase superfamily, is targeted to nucleoli. Virology 2011, 417, 334–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, M.; Seo, E.J.; Dever, T.E. Variola virus E3L Zalpha domain, but not its Z-DNA binding activity, is required for PKR inhibition. RNA 2014, 20, 214–227. [Google Scholar] [CrossRef] [Green Version]
- Langland, J.O.; Jacobs, B.L. Inhibition of PKR by vaccinia virus: Role of the N- and C-terminal domains of E3L. Virology 2004, 324, 419–429. [Google Scholar] [CrossRef] [Green Version]
- Szczerba, M.; Subramanian, S.; Trainor, K.; McCaughan, M.; Kibler, K.V.; Jacobs, B.L. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines 2022, 10, 235. [Google Scholar] [CrossRef]
- Rojas, M.; Vasconcelos, G.; Dever, T.E. An eIF2alpha-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2alpha. Proc. Natl. Acad. Sci. USA 2015, 112, E3466–E3475. [Google Scholar] [CrossRef] [Green Version]
- Denzler, K.L.; Schriewer, J.; Parker, S.; Werner, C.; Hartzler, H.; Hembrador, E.; Huynh, T.; Holechek, S.; Buller, R.M.; Jacobs, B.L. The attenuated NYCBH vaccinia virus deleted for the immune evasion gene, E3L, completely protects mice against heterologous challenge with ectromelia virus. Vaccine 2011, 29, 9691–9696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denzler, K.L.; Rice, A.D.; MacNeill, A.L.; Fukushima, N.; Lindsey, S.F.; Wallace, G.; Burrage, A.M.; Smith, A.J.; Manning, B.R.; Swetnam, D.M.; et al. The NYCBH vaccinia virus deleted for the innate immune evasion gene, E3L, protects rabbits against lethal challenge by rabbitpox virus. Vaccine 2011, 29, 7659–7669. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahman, M.J.; Tazi, L.; Haller, S.L.; Rothenburg, S. Crocodilepox Virus Protein 157 Is an Independently Evolved Inhibitor of Protein Kinase R. Viruses 2022, 14, 1564. https://doi.org/10.3390/v14071564
Rahman MJ, Tazi L, Haller SL, Rothenburg S. Crocodilepox Virus Protein 157 Is an Independently Evolved Inhibitor of Protein Kinase R. Viruses. 2022; 14(7):1564. https://doi.org/10.3390/v14071564
Chicago/Turabian StyleRahman, M. Julhasur, Loubna Tazi, Sherry L. Haller, and Stefan Rothenburg. 2022. "Crocodilepox Virus Protein 157 Is an Independently Evolved Inhibitor of Protein Kinase R" Viruses 14, no. 7: 1564. https://doi.org/10.3390/v14071564