
In Vitro Techniques and Measurements of Phage Characteristics That Are Important for Phage Therapy Success



Abstract
:1. Introduction
1.1. What Is a Phage?
1.2. What Is Phage Virulence?
1.3. The Challenge
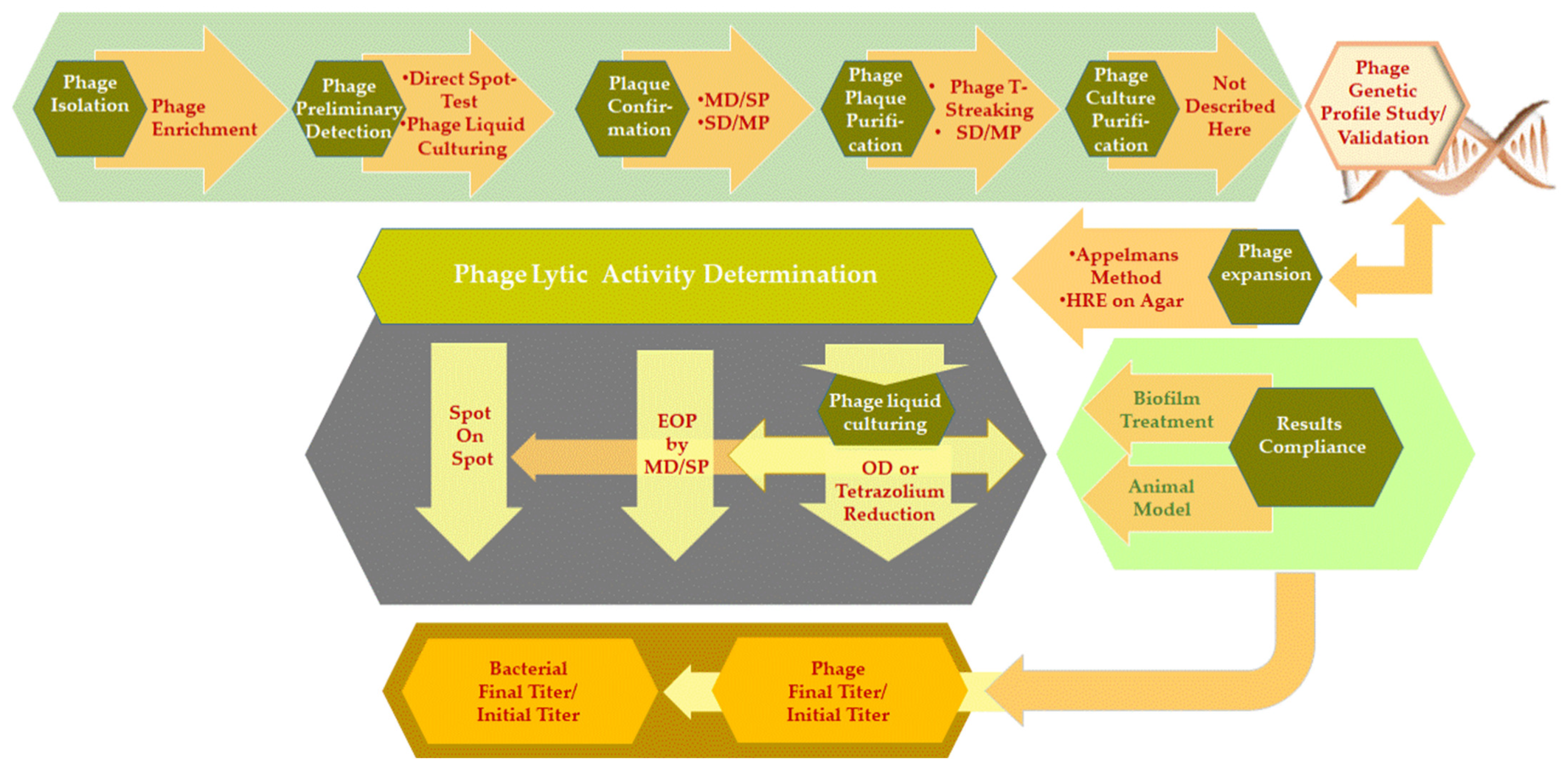
2. In Vitro and In Vivo Phage Detection and Phage Activity Testing
2.1. Phage Isolation Enrichment Method and Bacteria Hooks
- Bacteria hooks with hosts covering the wide range of receptors needed to hook the largest variety of potential phages. This requires having a readily available panel of strains with known genetic profiles. Every newly isolated phage can be further studied, e.g., to determine its biology;
- Bacteria hooks of particular interest can be included. In this case, bacterial strains are selected based on specific features such as antibiotic resistance, and it is not necessary to have an exhaustive list of characteristics or to know their genomic profile. The strains could be objects of further scientific study.
- Two times [35] or ten times [36] concentrated broth medium is typically added to the phage-sampling source to ensure sufficient nutrition. When using large sampling volumes, it is rational to use more concentrated (up to 20 times) broth media that will generate less volume of the end product, which makes it easier and safer when handling infectious material;
- It is preferable not to centrifuge/filter the sampling source, unless it contains large contaminants and/or components that will interfere with the incubation process. It is assumed that conditions close to those in the natural source environment will facilitate phage/bacteria interactions and the isolation of phages;
- Using lower temperatures (25–28 °C) than those routinely used in clinical microbiology (30–37 °C) [35,36] and longer incubation times, for instance 24 h (where commonly 4–6 h is enough for phage propagation in liquid media), are more favorable for PE. However, long incubation periods could also have an adverse effect on phage particles. Because the ratio of phage emergence to bacteria (those initially present in the sample and the added bacteria hooks) in the enrichment propagation mixture is not preliminary determined as obtaining consistent lysis without early (e.g., <24 h) phage-resistant bacterial mutant growth or phage antagonistic activity. In addition, some bacterial products could interfere with phage propagation or the demonstration of phage activity;
- Using 96- or 384-well microtiter plates for the incubation of a large number of inoculums of bacteria hooks is more convenient. The bacterial suspensions are collected from each well using a multichannel pipette (Appendix A, Figure A1);
- After incubation, the potential phage lysate (PL) is centrifuged and filtered. There is no necessity for the use of chloroform, as this could reduce the viruses’ infectivity [39] or inactivate some phages [16] and could also lead to the induction of temperate phages [40]. Using chloroform is a tradition that dates back to the time when bacterial filters were not available, and the procedure itself was not enough to ensure absolute removal of bacterial contamination. Adding the right amount (0.5–2% v/v) of chloroform to PL at +4 °C (temperature shock) kills the remaining intact bacterial cells, including lyrically phage-infected bacteria, and could thus result in substantially increased phage titers [16]. Chloroform was also used for the medium term (3–12 months) storage of phage stocks, as it prevented bacterial growth [41]. In addition to the obvious laboratory personnel safety issues (hazardous chemicals), it is not recommended to use chloroform for phage preparations that will be used in clinical treatments;
- The obtained PL could be used further as the second source for another enrichment BP with different bacteria hooks.
Phage Detection—Preliminary Tests
- (i)
- The “direct spot test” (here, we call it a technique): in which only one dilution of the phage lysate is spotted on bacteria grown directly on solid agar. It is described below;
- (ii)
- (iii)
- The “lysis profile assay” [21] or, as we call it here, “phage liquid culturing” (PLC) method implies the liquid culture of phage/bacteria mixtures at specific dilution(s) in microtiter plates for the determination of phage susceptibility. As many as 5- to 10-fold greater numbers of bacterial test strains could be considered per microtiter plate, as compared to the conventional “spot tests” performed on petri dishes of different sizes and shapes [28]. This results in reduced hands-on time and fewer consumables.
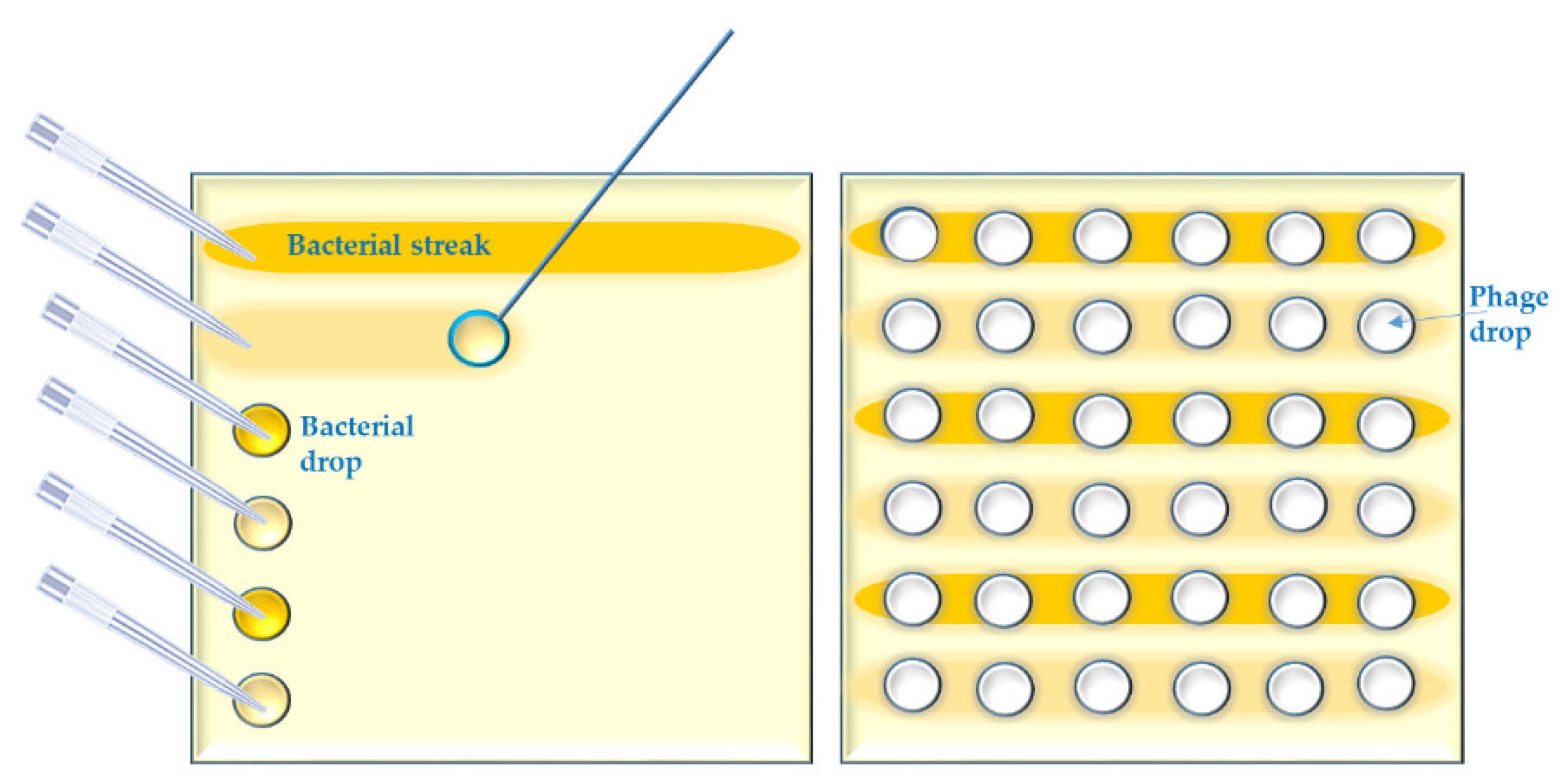
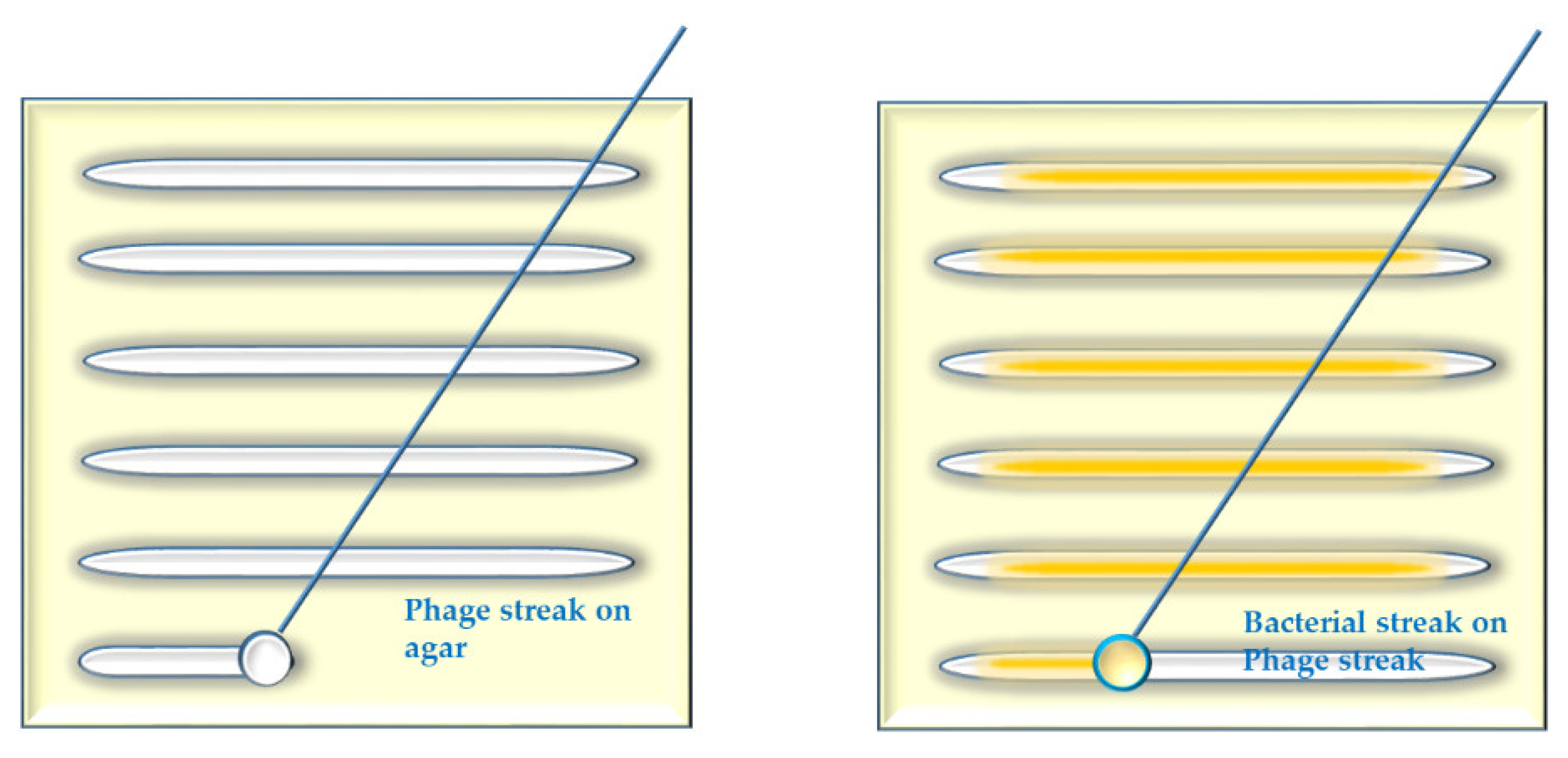
- Several parallel streaks (“streak assay” [36,46]) of bacterial suspension(s) of particular dilution(s) are made using disposable loops (Appendix A, Figure A2). Phage lysate(s) are applied as spots on the bacterial streaks (we call it “spot-on-streak” to differentiate from the other techniques);
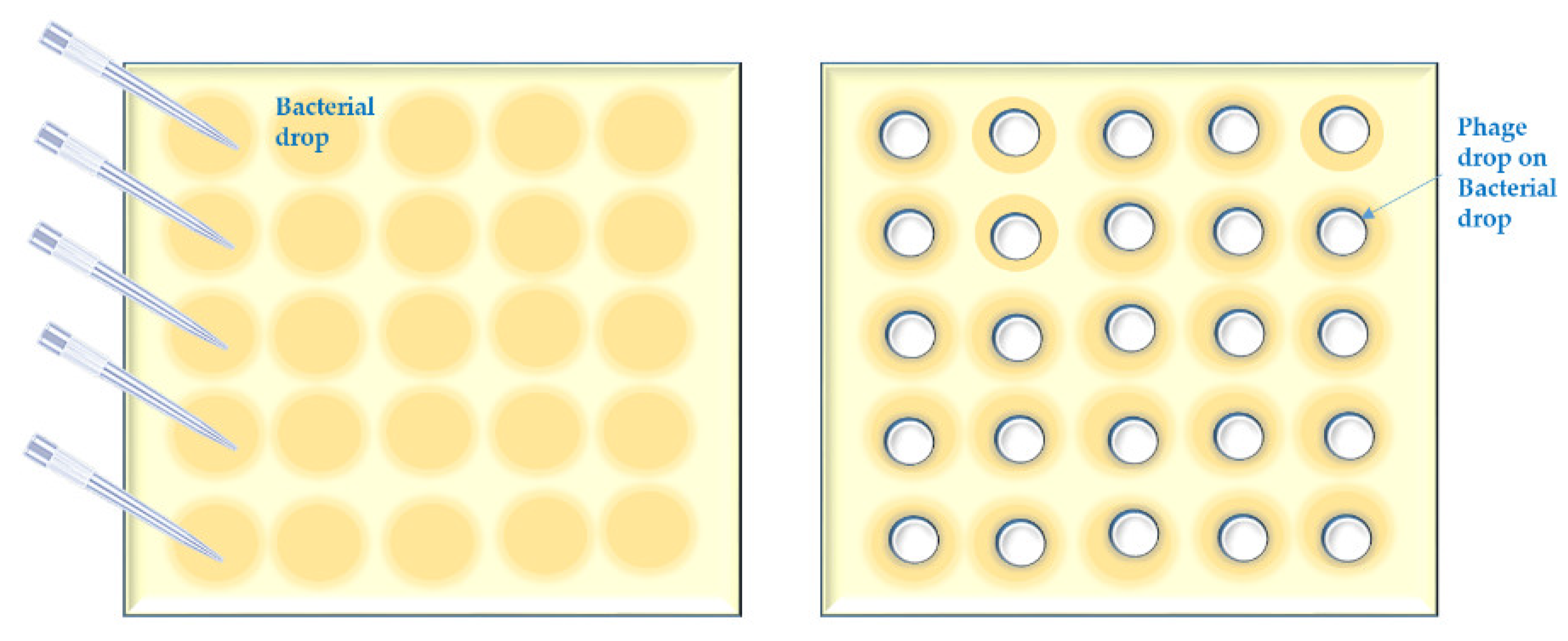
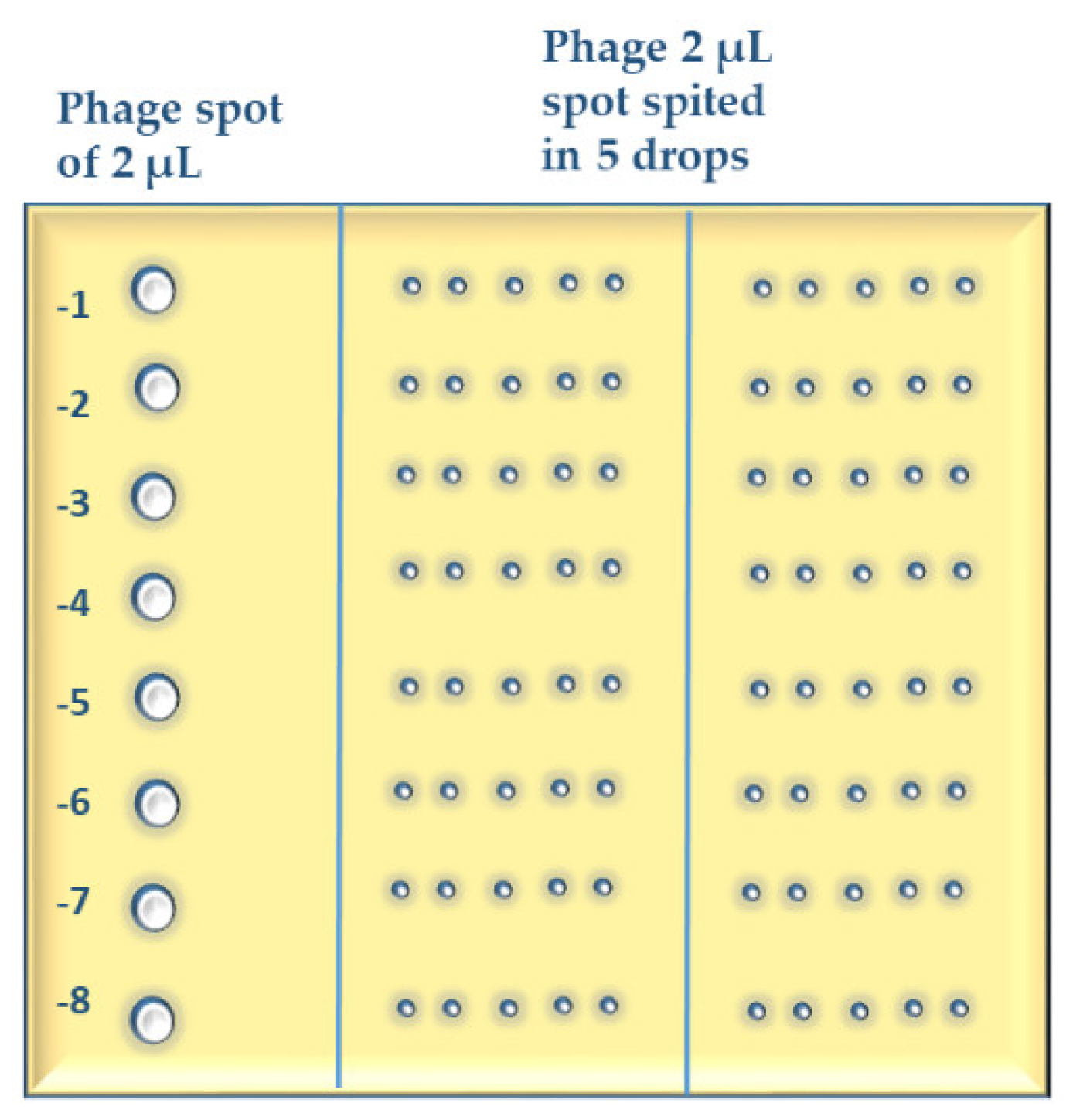
- Bacterial suspensions are simply spotted [47] in a grid. Phage lysate(s) are applied as spots (we call it “spot-on-spot”) (Appendix A, Figure A3);
- Bacterial suspensions are directly streaked on streaks of phages made on solid agar [48] (we call it “streak-on-streak”) (Appendix A, Figure A4).
2.2. Confirmatory Test for Phage Activity Detection/Enumeration—Plaque Formation
- Confirmation of plaque formation;
- Study of plaque morphology;
- Enumeration (determination of pfu/mL) of phages.
- Phage differentiation/selection;
- Plaque purification;
- Phage virulence/lysogeny evaluation procedures.
2.2.1. Double Agar Layer (DAL) Method
- Plaque diameter;
- Level of transparency/turbidity of the plaques;
- Halo formation and size;
- Motility.
2.2.2. Plaque Purification
- The distance between the plaques (well isolated discrete plaques);
- Different dilutions of phage lysate are applied;
- A certain number of passaging rounds are performed (3–5 final confirmation rounds);
- Several bacterial host bacterial strains are used;
- Several growth media are used.
2.2.3. Bacteria Kits for the Study of Phage Host Range and Efficiency of Plating (EOP)
2.3. Phage Liquid Culturing Method and the Translation of Results
- Phage enumeration with phage titer expressed as a dilution factor;
- Estimation of the multiplicity of infection (MOI) [10], i.e., the ratio of phages to bacteria, for instance, to set the initial phage/bacterium inoculates for in vitro/vivo studies;
- Evaluation of host range and lytic activity [17];
- Expansion of host range after multiple passaging.
Host Range Expansion (HRE)
3. Discussion
3.1. Bacterial Population and Infection Locus Consistency
3.2. Phage-Bacteria Ratio
3.3. Phage Mixtures
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A
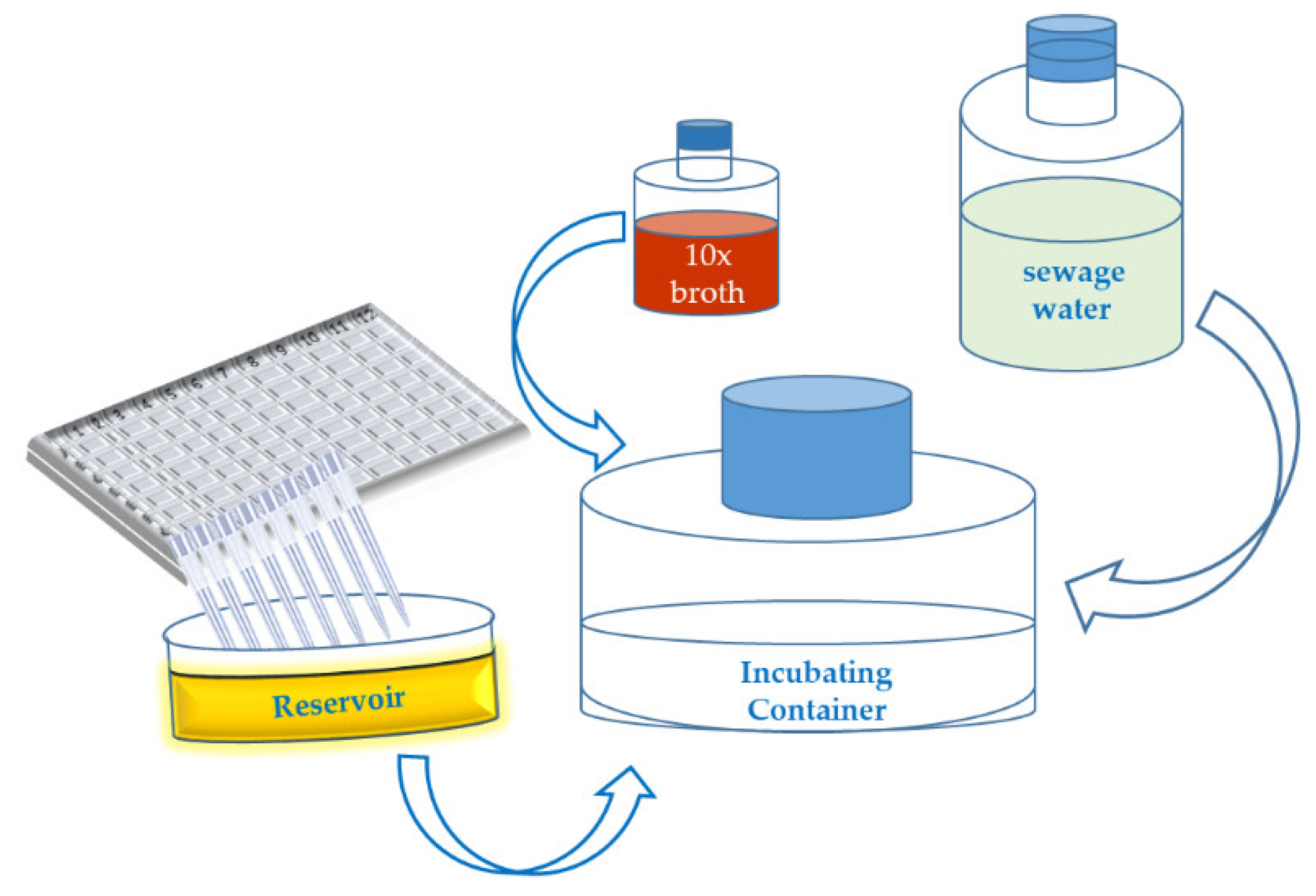
Appendix A.1. Short Outline of the “Phage Isolation Enrichment” Method
- 1.1
- Culture the bacterial strains in 96- or 384-well microtiter plates overnight at an appropriate temperature, in a suitable culture medium.
- 1.2
- Collect 200 (40) µL of each of bacterial suspension from each well of the 96 (384)-well microtiter plates (19.2 (15.36) mL in total correspondingly) and transfer the liquid to a sterile reservoir using a multichannel pipette.
- 1.3
- Add the following ingredients to a sterile container (flask):
- 360 (288) mL of sewage water.
- 40 (32) mL of 10× concentrated culture medium (broth).
- 19.2 (15.36) mL mixture of the bacterial suspensions in the reservoir.
- 1.4
- Incubate the container at 25–28 °C for 18–24 h.
- 1.5
- Centrifuge the (potential) phage lysate at 6000× g for 30 min.
- 1.6
- Filtrate the (potential) phage lysate using a 0.45 µm syringe filter.
- 1.7
- Store the supernatant at 4 °C.

Appendix A.2. Short Outline of the “Spot on Streak” Method
- 2.1
- Make dilutions of the bacterial suspensions in a 96-well-microtiter plate including the following two dilutions: a low concentration containing 1.0 × 104 cfu/mL and an average one containing 1.0 × 107 cfu/mL.
- 2.2
- Apply a drop (20 µL) of each bacterial suspension in the first column of a grid on a square petri dish containing a suitable agar medium, using a multichannel pipette; then roll down each drop to the end of the grid row by using the same pipette and tips or separate disposable loops. Let the bacterial streak dry up in a Biosafety Cabinet (BSC).
- 2.3
- Distribute the phage lysates in a 96-well-microtiter plate or another segmented reservoir according to their foreseen outline on the test agar plate grids. Spot 10 µL of phage lysates on the bacterial streaks in a vertical direction by multichannel pipet.
- 2.4
- Let the spots dry up in a BSC and then incubate the test plates upside down at a temperature of 25–28 °C (which should be lower than the standard incubation temperature for the considered bacterial strains) for 18 h.

Appendix A.3. Short Outline of the “Spot on Spot” Method
- 3.1
- Repeat the first step of the “spot-test on streak” method.
- 3.2
- Spot 10 µL of the bacterial suspensions in the first column of the grid. Let the bacterial spot dry up in a BSC.
- 3.3
- Spot 5 µL of phage lysate over the bacterial spot.
- 3.4
- Repeat step 2.4. of the “spot on bacterial streak” method.

Appendix A.4. Short Outline of the “Streak on Streak” Method
- 4.1
- Apply phage lysate drops (20 µL) in the first column of a grid on a square petri dish containing a suitable agar medium, using a multichannel pipette; then roll down each drop to the end of the grid row by using the same pipette and tips or separate disposable loops. Don’t allow phage streaks to dry up before bacterial suspensions are applied.
- 4.2
- Streak 10 µL of bacterial suspensions over the phage streaks. Let the bacteria/phage streaks dry up in a BSC.
- 4.3
- Repeat step 2.4. of the “spot on streak” method.

Appendix A.5. Short Outline of the MD/SP (Multiple Dilutions on Single Plates) Method
- 5.1
- Make ten-fold serial dilutions of phage lysate(s) in 96-well microtiter plates (add 20 µL of phage suspension to 180 µL of phosphate buffered saline) typically up to 10−8.
- 5.2
- Mix 300 µL of bacterial suspension of an OD that is preliminary adjusted for each host strain or species with up to 8 mL of molten soft agar (0.7% or 0.8% suitable agar 46 °C) in a 15 mL tube and pour the mixture onto pre-prepared square petri dishes with 1.5% agar medium. Use 0.8% soft agar for phages that form large plaques. Let the plates dry up for 10–15 min in a BSC.
- 5.3
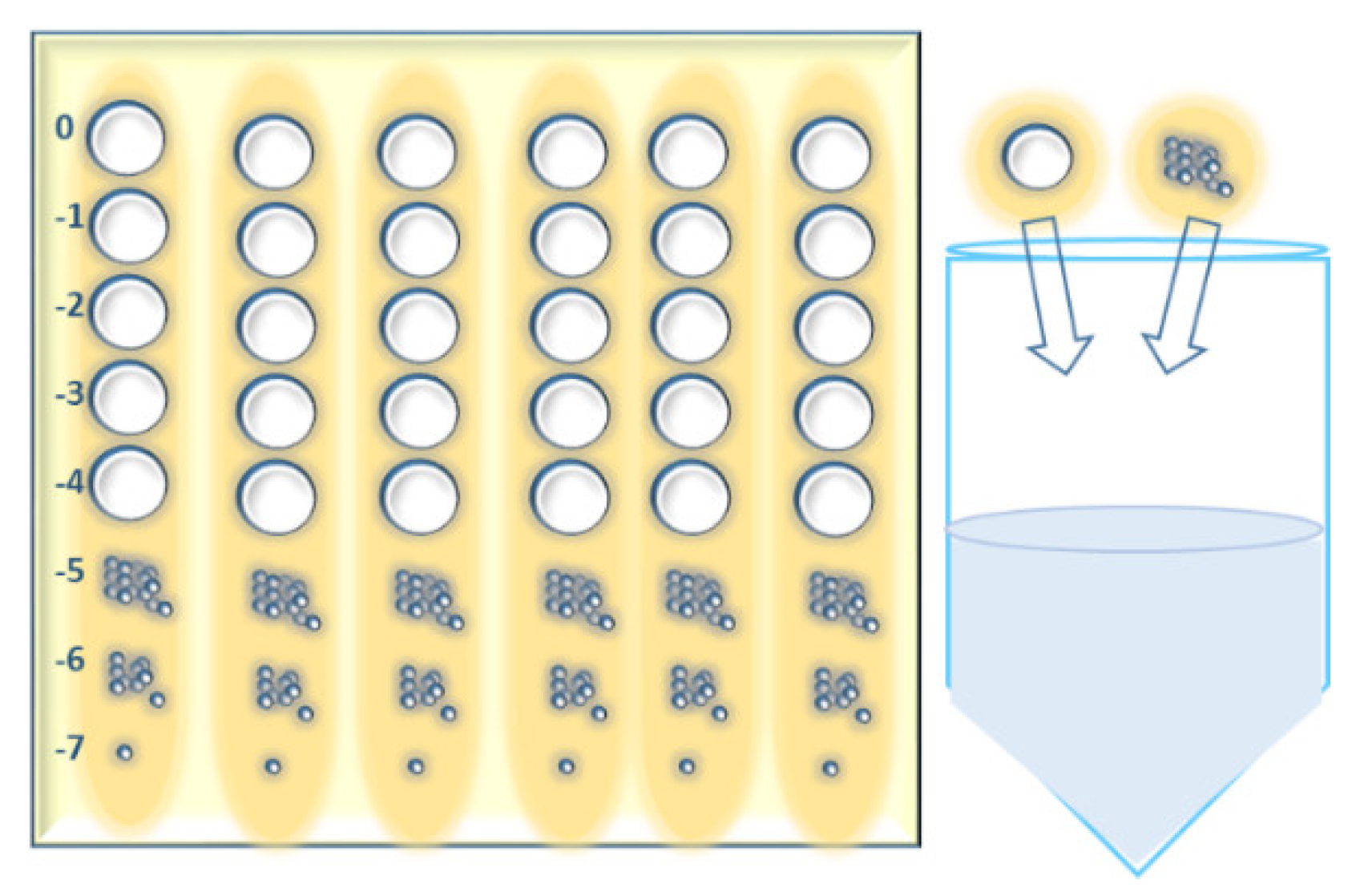
- Spot 2 µL of each phage dilution onto the soft agar surface across the column of the plate grid (six columns on a square petri dish) using a multichannel pipette. Make three repetitions of each test phage. In case of phages with large plaques, make a three-column grid on a square petri dish and split the 2-µL-spot in 4 smaller drops while applying on the agar surface.
- 5.4
- Use standard phage dilutions (with known titer), on each test plate (whenever possible) as control for the titration.
- 5.5
- Let the test plates dry in a BSC and incubate them upside down at 28–32 °C (depending on the host bacteria) for 18–24 h.
- 5.6
- After incubation, calculate the average number of plaques for the different dilutions and repetitions and multiply them by 500 to obtain the number of plaques in 1 mL. The phage titer (pfu/mL) is the number of plaques in 1 mL multiplied by the reciprocal of dilution.

Appendix A.6. Short Outline of “Host Range Expansion (HRE) on Agar” Method
- 6.1
- Make phage mixture dilutions as described in the MD/SP method (step 5.1.).
- 6.2
- Make bacterial streaks lines of 30 µL as described in the “spot on streak” method (steps 2.1.–2.2.). Six lines in total are made on a square petri dish.
- 6.3
- Spot 10 µL of each phage mixture dilution (from zero dilution to 10−7) lengthways on the bacterial lines.
- 6.4
- Repeat step 2.4. of the “spot on streak” method.
- 6.5
- After incubation, cut out all agar zones with different clearings (from clear to separate plaques). If there is no sign of phage activity on a particular strain, cut out the agar from the zero dilution zone only.
- 6.6
- Collect all agar cuts in one container and add a volume of phosphate buffered saline corresponding to 3–5 mL per agar cut.
- 6.7
- Stir the container with its content for 1–1.5 h at 400 min−1 and then centrifuge at 6000× g for 30 min.
- 6.8
- Filtrate the supernatant using a 0.45 µm syringe filter.
- 6.9
- Repeat the passaging rounds until the expected phage host-range extension is obtained.

References
- Myelnikov, D. Creature features: The lively narratives of bacteriophages in Soviet biology and medicine. Notes Rec. R. Soc. J. Hist. Sci. 2020, 74, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Summers, W.C. Félix Hubert d’Herelle (1873–1949): History of a scientific mind. Bacteriophage 2016, 6, e1270090. [Google Scholar] [CrossRef]
- Abedon, S.T.; Danis-Wlodarczyk, K.M.; Alves, D.R. Phage Therapy in the 21st Century: Is There Modern, Clinical Evidence of Phage-Mediated Efficacy? Pharmaceuticals 2021, 14, 1157. [Google Scholar] [CrossRef] [PubMed]
- Witzany, G. What does communication of phages mean? In Biocommunication of Phages; Witzany, G., Ed.; Springer: Berlin/Heidelberg, Germany, 2020. [Google Scholar] [CrossRef]
- Pirnay, J.-P.; De Vos, D.; Verbeken, G.; Merabishvili, M.; Chanishvili, N.; Vaneechoutte, M.; Zizi, M.; Laire, G.; Lavigne, R.; Huys, I.; et al. The Phage Therapy Paradigm: Prêt-à-Porter or Sur-mesure? Pharm. Res. 2011, 28, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Chen, I.A. Phage engineering and the evolutionary arms race. Curr. Opin. Biotechnol. 2021, 68, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Weinbauer, M.G. Ecology of prokaryotic viruses. FEMS Microbiol. Rev. 2004, 28, 127–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedon, S.T.; García, P.; Mullany, P.; Aminov, R. Editorial: Phage Therapy: Past, Present and Future. Front. Microbiol. 2017, 8, 981. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T.; Danis-Wlodarczyk, K.M.; Wozniak, D.J.; Sullivan, M.B. Improving Phage-Biofilm In Vitro Experimentation. Viruses 2021, 13, 1175. [Google Scholar] [CrossRef]
- Monk, A.B.; Rees, C.D.; Barrow, P.; Hagens, S.; Harper, D.R. Bacteriophage applications: Where are we now? Lett. Appl. Microbiol. 2010, 51, 363–369. [Google Scholar] [CrossRef]
- Casadevall, A.; Pirofski, L. Host-Pathogen Interactions: The Attributes of Virulence. J. Infect. Dis. 2001, 184, 337–344. [Google Scholar] [CrossRef]
- Hobbs, Z.; Abedon, S.T. Diversity of phage infection types and associated terminology: The problem with ‘Lytic or lysogenic’. FEMS Microbiol. Lett 2016, 363, fnw047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storms, Z.J.; Teel, M.R.; Mercurio, K.; Sauvageau, D. The Virulence Index: A Metric for Quantitative Analysis of Phage Virulence. Phage 2020, 1, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.D.; Johnson, R.P.; Xu, Y.; McAllister, T.A.; Sharma, R.; Louie, M.; Stanford, K. Host range and lytic capability of four bacteriophages against bovine and clinical human isolates of Shiga toxin-producing Escherichia coli O157:H7. J. Appl. Microbiol. 2009, 107, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Hyman, P. Phage Choice, Isolation, and Preparation for Phage Therapy. Curr. Pharm. Biotechnol. 2010, 11, 2–14. [Google Scholar] [CrossRef]
- Hyman, P. Phages for Phage Therapy: Isolation, Characterization, and Host Range Breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Wahab, L.; Gill, J. Development and Validation of a Microtiter Plate-Based Assay for Determination of Bacteriophage Host Range and Virulence. Viruses 2018, 10, 189. [Google Scholar] [CrossRef] [Green Version]
- Mirzaei, M.K.; Nilsson, A.S. Correction: Isolation of Phages for Phage Therapy: A Comparison of Spot Tests and Efficiency of Plating Analyses for Determination of Host Range and Efficacy. PLoS ONE 2015, 10, e0127606. [Google Scholar] [CrossRef] [Green Version]
- Skurnik, M. Can Bacteriophages Replace Antibiotics? Antibiotics 2022, 11, 575. [Google Scholar] [CrossRef]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S. Phage Therapy in Clinical Practice: Treatment of Human Infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef]
- Danis-Wlodarczyk, K.M.; Cai, A.; Chen, A.; Gittrich, M.R.; Sullivan, M.B.; Wozniak, D.J.; Abedon, S.T. Friends or Foes? Rapid Determination of Dissimilar Colistin and Ciprofloxacin Antagonism of Pseudomonas aeruginosa Phages. Pharmaceuticals 2021, 14, 1162. [Google Scholar] [CrossRef]
- Lindberg, H.M.; McKean, K.A.; Wang, I.-N. Phage fitness may help predict phage therapy efficacy. Bacteriophage 2014, 4, e964081. [Google Scholar] [CrossRef] [PubMed]
- Rakhuba, D.V.; Kolomiets, E.I.; Dey, E.S.; Novik, G.I. Bacteriophage receptors, mechanisms of phage adsorption and penetration into host cell. Pol. J. Microbiol. 2010, 59, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Santos, S.B.; Carvalho, C.; Azeredo, J.; Ferreira, E.C. Correction: Population Dynamics of a Salmonella Lytic Phage and Its Host: Implications of the Host Bacterial Growth Rate in Modelling. PLoS ONE 2015, 10, e0136007. [Google Scholar] [CrossRef] [PubMed]
- Payne, R. Phage therapy: The peculiar kinetics of self-replicating pharmaceuticals. Clin. Pharmacol. Ther. 2000, 68, 225–230. [Google Scholar] [CrossRef] [Green Version]
- Cairns, B.J.; Timms, A.R.; Jansen, V.A.A.; Connerton, I.F.; Payne, R.J.H. Quantitative Models of In Vitro Bacteriophage–Host Dynamics and Their Application to Phage Therapy. PLoS Pathog. 2009, 5, e1000253. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.L.; Sieo, C.C.; Tan, W.S.; Abdullah, N.; Hair-Bejo, M.; Abu, J.; Ho, Y.W. Evaluation of a lytic bacteriophage, Φ st1, for biocontrol of Salmonella enterica serovar Typhimurium in chickens. Int. J. Food Microbiol. 2016, 172, 92–101. [Google Scholar] [CrossRef]
- Weber-Dąbrowska, B.; Jończyk-Matysiak, E.; Żaczek, M.; Łobocka, M.; Łusiak-Szelachowska, M.; Górski, A. Bacteriophage Procurement for Therapeutic Purposes. Front. Microbiol. 2016, 7, 1177. [Google Scholar] [CrossRef] [Green Version]
- Sargeant, K. Large-Scale Bacteriophage Production. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 1970; Volume 13, pp. 121–137. [Google Scholar] [CrossRef]
- Harper, D. Criteria for Selecting Suitable Infectious Diseases for Phage Therapy. Viruses 2018, 10, 177. [Google Scholar] [CrossRef] [Green Version]
- Forti, F.; Roach, D.R.; Cafora, M.; Pasini, M.E.; Horner, D.S.; Fiscarelli, E.V.; Rossitto, M.; Cariani, L.; Briani, F.; Debarbieux, L.; et al. Design of a Broad-Range Bacteriophage Cocktail That Reduces Pseudomonas aeruginosa Biofilms and Treats Acute Infections in Two Animal Models. Antimicrob. Agents Chemother. 2018, 62, e02573-17. [Google Scholar] [CrossRef] [Green Version]
- Krut, O.; Bekeredjian-Ding, I. Contribution of the Immune Response to Phage Therapy. J. Immunol. 2018, 200, 3037–3044. [Google Scholar] [CrossRef]
- Kropinski, A.M.; Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of bacteriophages by double agar overlay plaque assay. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 69–76. [Google Scholar] [CrossRef]
- Cooper, C.J.; Denyer, S.P.; Maillard, J.-Y. Rapid and quantitative automated measurement of bacteriophage activity against cystic fibrosis isolates of Pseudomonas aeruginosa: Rapid quantitative screening of phage activity. J. Appl. Microbiol. 2011, 110, 631–640. [Google Scholar] [CrossRef]
- Twest, R.; Kropinski, A.M. Bacteriophage Enrichment from Water and Soil. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 15–21. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.-P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-Controlled Small-Scale Production of a Well-Defined Bacteriophage Cocktail for Use in Human Clinical Trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Mathieu, J.; Li, M.; Dai, Z.; Alvarez, P.J.J. Isolation of Polyvalent Bacteriophages by Sequential Multiple-Host Approaches. Appl. Environ. Microbiol. 2016, 82, 808–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olsen, N.S.; Hendriksen, N.B.; Hansen, L.H.; Kot, W. A New High-Throughput Screening Method for Phages: Enabling Crude Isolation and Fast Identification of Diverse Phages with Therapeutic Potential. Phage 2020, 1, 137–148. [Google Scholar] [CrossRef]
- Kauffman, K.M.; Polz, M.F. Streamlining standard bacteriophage methods for higher throughput. MethodsX 2018, 5, 159–172. [Google Scholar] [CrossRef]
- Willner, D.; Furlan, M.; Schmieder, R.; Grasis, J.A.; Pride, D.T.; Relman, D.A.; Angly, F.E.; McDole, T.; Mariella, R.P.; Rohwer, F.; et al. Metagenomic detection of phage-encoded platelet-binding factors in the human oral cavity. Proc. Natl. Acad. Sci. USA 2010, 108, 4547–4553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.-K.; Liu, Y.-L.; Hu, A.; Chang, K.-C.; Lin, N.-T.; Lai, M.-J.; Tseng, C.-C. Potential of bacteriophage ΦAB2 as an environmental biocontrol agent for the control of multidrug-resistant Acinetobacter baumannii. BMC Microbiol. 2013, 13, 154. [Google Scholar] [CrossRef] [Green Version]
- De Jonge, P.A.; Nobrega, F.L.; Brouns, S.J.J.; Dutilh, B.E. Molecular and Evolutionary Determinants of Bacteriophage Host Range. Trends Microbiol. 2019, 27, 51–63. [Google Scholar] [CrossRef]
- Amorim, L.R.P.; Silva, J.G.L.; Gibbs, P.A.; Teixeira, P.C. Application of an Impedimetric Technique for the Detection of Lytic Infection of Salmonella spp. By Specific Phages. Int. J. Microbiol. 2009, 2009, 259456. [Google Scholar] [CrossRef] [Green Version]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2004; p. 528. ISBN 9780429208720. [Google Scholar] [CrossRef]
- Haines, M.E.K.; Hodges, F.E.; Nale, J.Y.; Mahony, J.; van Sinderen, D.; Kaczorowska, J.; Alrashid, B.; Akter, M.; Brown, N.; Sauvageau, D.; et al. Analysis of Selection Methods to Develop Novel Phage Therapy Cocktails Against Antimicrobial Resistant Clinical Isolates of Bacteria. Front. Microbiol. 2021, 12, 613529. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.D. Bacteriophages; Interscience Publishers: New York, NY, USA, 1959. [Google Scholar]
- Saussereau, E.; Vachier, I.; Chiron, R.; Godbert, B.; Sermet, I.; Dufour, N.; Pirnay, J.-P.; De Vos, D.; Carrié, F.; Molinari, N.; et al. Effectiveness of bacteriophages in the sputum of cystic fibrosis patients. Clin. Microbiol. Infect. 2014, 20, O983–O990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betts, A.; Vasse, M.; Kaltz, O.; Hochberg, M.E. Back to the future: Evolving bacteriophages to increase their effectiveness against the pathogen P seudomonas aeruginosa PAO 1. Evol. Appl. 2013, 6, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Letarov, A.V.; Kulikov, E.E. Determination of the bacteriophage host range: Culture-based approach. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Springer: New York, NY, USA, 2018; Volume 1693, pp. 75–84. [Google Scholar] [CrossRef]
- Hyman, P.; Abedon, S.T. Bacteriophage host range and Bacterial resistance. In Advances in Applied Microbiology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 70, pp. 217–248. [Google Scholar] [CrossRef]
- Abedon, S.T.; Yin, J. Bacteriophage plaques: Theory and analysis. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 161–174. [Google Scholar] [CrossRef]
- Kutter, E. Phage Host Range and Efficiency of Plating. In Bacteriophages; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501, pp. 141–149. [Google Scholar] [CrossRef]
- Mazzocco, A.; Waddell, T.E.; Lingohr, E.; Johnson, R.P. Enumeration of Bacteriophages Using the Small Drop Plaque Assay System. In Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; Volume 501. [Google Scholar] [CrossRef]
- Abedon, S.T. Lysis from without. Bacteriophage 2011, 1, 46–49. [Google Scholar] [CrossRef]
- Kakabadze, E.; Makalatia, K.; Grdzelishvili, N.; Bakuradze, N.; Goderdzishvili, M.; Kusradze, I.; Phoba, M.-F.; Lunguya, O.; Lood, C.; Lavigne, R.; et al. Selection of Potential Therapeutic Bacteriophages that Lyse a CTX-M-15 Extended Spectrum β-Lactamase Producing Salmonella enterica Serovar Typhi Strain from the Democratic Republic of the Congo. Viruses 2018, 10, 172 . [Google Scholar] [CrossRef] [Green Version]
- Sillankorva, S. Isolation of Bacteriophages for Clinically Relevant Bacteria. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Springer: New York, NY, USA, 2018; Volume 1693, pp. 23–30. [Google Scholar] [CrossRef]
- Kusradze, I.; Karumidze, N.; Rigvava, S.; Dvalidze, T.; Katsitadze, M.; Amiranashvili, I.; Goderdzishvili, M. Characterization and Testing the Efficiency of Acinetobacter baumannii Phage vB-GEC_Ab-M-G7 as an Antibacterial Agent. Front. Microbiol. 2016, 7, 1590. [Google Scholar] [CrossRef] [Green Version]
- Uchiyama, J.; Takemura, I.; Satoh, M.; Kato, S.; Ujihara, T.; Akechi, K.; Matsuzaki, S.; Daibata, M. Improved Adsorption of an Enterococcus faecalis Bacteriophage ΦEF24C with a Spontaneous Point Mutation. PLoS ONE 2011, 6, e26648. [Google Scholar] [CrossRef]
- Merabishvili, M.; Pirnay, J.-P.; De Vos, D. Guidelines to Compose an Ideal Bacteriophage Cocktail. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Springer: New York, NY, USA, 2018; Volume 1693, pp. 99–110. [Google Scholar] [CrossRef]
- Burrowes, B.; Molineux, I.; Fralick, J. Directed in Vitro Evolution of Therapeutic Bacteriophages: The Appelmans Protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef] [Green Version]
- Pirnay, J.-P.; Blasdel, B.G.; Bretaudeau, L.; Buckling, A.; Chanishvili, N.; Clark, J.R.; Corte-Real, S.; Debarbieux, L.; Dublanchet, A.; De Vos, D.; et al. Quality and Safety Requirements for Sustainable Phage Therapy Products. Pharm. Res. 2015, 32, 2173–2179. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N. Bacteriophages as Therapeutic and Prophylactic Means: Summary of the Soviet and Post Soviet Experiences. Curr. Drug Deliv. 2016, 13, 309–323. [Google Scholar] [CrossRef]
- Christiansen, B.; Johnsen, M.G.; Stenby, E.; Vogensen, F.K.; Hammer, K. Characterization of the lactococcal temperate phage TP901-1 and its site-specific integration. J. Bacteriol. 1994, 176, 1069–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, J.J.; Gill, J.J. The habits of highly effective phages: Population dynamics as a framework for identifying therapeutic phages. Front. Microbiol. 2014, 5, 618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stockdale, S.; Mahony, J.; Courtin, P.; Chapot-Chartier, M.-P.; van Pijkeren, J.-P.; Britton, R.A.; Neve, H.; Heller, K.J.; Aideh, B.; Vogensen, F.; et al. The Lactococcal Phages Tuc2009 and TP901-1 Incorporate Two Alternate Forms of Their Tail Fiber into Their Virions for Infection Specialization. J. Biol. Chem. 2013, 288, 5581–5590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kropinski, A.M.; Waddell, T.; Meng, J.; Franklin, K.; Ackermann, H.-W.; Ahmed, R.; Mazzocco, A.; Yates, J.; Lingohr, E.J.; Johnson, R.P. The host-range, genomics and proteomics of Escherichia coli O157:H7 bacteriophage rV5. Virol. J. 2013, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Raya, R.R.; Varey, P.; Oot, R.A.; Dyen, M.R.; Callaway, T.R.; Edrington, T.S.; Kutter, E.M.; Brabban, A.D. Isolation and Characterization of a New T-Even Bacteriophage, CEV1, and Determination of Its Potential To Reduce Escherichia coli O157:H7 Levels in Sheep. Appl. Environ. Microbiol. 2006, 72, 6405–6410. [Google Scholar] [CrossRef] [Green Version]
- Vandersteegen, K.; Kropinski, A.M.; Nash, J.H.E.; Noben, J.-P.; Hermans, K.; Lavigne, R. Romulus and Remus, Two Phage Isolates Representing a Distinct Clade within the Twortlikevirus Genus, Display Suitable Properties for Phage Therapy Applications. J. Virol. 2013, 87, 3237–3247. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.D.; Stanford, K.; Kropinski, A.M.; Ackermann, H.-W.; Johnson, R.P.; She, Y.-M.; Ahmed, R.; Villegas, A.; McAllister, T.A. Genomic, Proteomic and Physiological Characterization of a T5-like Bacteriophage for Control of Shiga Toxin-Producing Escherichia coli O157:H7. PLoS ONE 2012, 7, e34585. [Google Scholar] [CrossRef] [Green Version]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.-J.; Bilocq, F.; De Vos, D.; Pirnay, J.-P.; Noben, J.-P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and Molecular Assessment of Bacteriophage ISP for the Control of Staphylococcus aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, W.N.; Haq, I.U.; Andleeb, S.; Qadri, I. Characterization of a virulent bacteriophage LK1 specific for Citrobacter freundii isolated from sewage water. J. Basic Microbiol. 2014, 54, 531–541. [Google Scholar] [CrossRef]
- Niu, Y.D.; McAllister, T.A.; Nash, J.; Kropinski, A.; Stanford, K. Four Escherichia coli O157:H7 Phages: A New Bacteriophage Genus and Taxonomic Classification of T1-Like Phages. PLoS ONE 2014, 9, e100426. [Google Scholar] [CrossRef] [Green Version]
- Henry, M.; Lavigne, R.; Debarbieux, L. Predicting In Vivo Efficacy of Therapeutic Bacteriophages Used To Treat Pulmonary Infections. Antimicrob. Agents Chemother. 2013, 57, 5961–5968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, S.I.; Kaelber, J.T.; Ma, L.; Trautner, B.W.; Ramig, R.F.; Maresso, A.W. Bacteriophages from ExPEC Reservoirs Kill Pandemic Multidrug-Resistant Strains of Clonal Group ST131 in Animal Models of Bacteremia. Sci. Rep. 2017, 7, 46151. [Google Scholar] [CrossRef] [PubMed]
- Estrella, L.A.; Quinones, J.; Henry, M.; Hannah, R.M.; Pope, R.K.; Hamilton, T.; Teneza-mora, N.; Hall, E.; & Biswajit, B. Characterization of novel Staphylococcus aureus lytic phage and defining their combinatorial virulence using the OmniLog® system. Bacteriophage 2016, 6, e1219440. [Google Scholar] [CrossRef] [Green Version]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and Use of Personalized Bacteriophage-Based Therapeutic Cocktails To Treat a Patient with a Disseminated Resistant Acinetobacter baumannii Infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [Green Version]
- LaVergne, S.; Hamilton, T.; Biswas, B.; Kumaraswamy, M.; Schooley, R.T.; Wooten, D. Phage Therapy for a Multidrug-Resistant Acinetobacter baumannii Craniectomy Site Infection. Open Forum Infect. Dis. 2018, 5, ofy064. [Google Scholar] [CrossRef] [Green Version]
- Konopacki, M.; Grygorcewicz, B.; Dołęgowska, B.; Kordas, M.; Rakoczy, R. PhageScore: A simple method for comparative evaluation of bacteriophages lytic activity. Biochem. Eng. 2020, 161, 107652. [Google Scholar] [CrossRef]
- Nale, J.Y.; Vinner, G.K.; Lopez, V.C.; Thanki, A.M.; Phothaworn, P.; Thiennimitr, P.; Garcia, A.; AbuOun, M.; Anjum, M.F.; Korbsrisate, S.; et al. An Optimized Bacteriophage Cocktail Can Effectively Control Salmonella in vitro and in Galleria mellonella. Front. Microbiol. 2021, 11, 609955. [Google Scholar] [CrossRef]
- Ebert, D. Experimental Evolution of Parasites. Science 1998, 282, 1432–1436. [Google Scholar] [CrossRef] [Green Version]
- Hall, A.R.; De Vos, D.; Friman, V.-P.; Pirnay, J.-P.; Buckling, A. Effects of Sequential and Simultaneous Applications of Bacteriophages on Populations of Pseudomonas aeruginosa In Vitro and in Wax Moth Larvae. Appl. Environ. Microbiol. 2012, 78, 5646–5652. [Google Scholar] [CrossRef] [Green Version]
- Poullain, V.; Gandon, S.; Brockhurst, M.A.; Buckling, A.; Hochberg, M.E. The evolution of specificity in evolving and coevolving antagonistic interactions between a bacteria and its phage. Evolution 2007, 62, 1–11. [Google Scholar] [CrossRef]
- Friman, V.-P.; Soanes-Brown, D.; Sierocinski, P.; Buckling, A.; Johansen, H.K.; Molin, S.; Merabishvili, M. Data from: Pre-adapting parasitic phages to a pathogen leads to increased pathogen clearance and lowered resistance evolution with Pseudomonas aeruginosa cystic fibrosis bacterial isolates. J. Evol. Biol. 2016, 29, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneumoniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef] [PubMed]
- Burrowes, B.; Harper, D.R.; Anderson, J.; McConville, M.; Enright, M.C. Bacteriophage therapy: Potential uses in the control of antibiotic-resistant pathogens. Expert Rev. Anti-infective Ther. 2011, 9, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Mapes, A.C.; Trautner, B.W.; Liao, K.S.; Ramig, R.F. Development of expanded host range phage active on biofilms of multi-drug resistant Pseudomonas aeruginosa. Bacteriophage 2016, 6, e1096995. [Google Scholar] [CrossRef] [Green Version]
- Rossitto, M.; Fiscarelli, E.V.; Rosati, P. Challenges and Promises for Planning Future Clinical Research Into Bacteriophage Therapy Against Pseudomonas aeruginosa in Cystic Fibrosis. An Argumentative Review. Front. Microbiol. 2018, 9, 775. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [Green Version]
- Bourdin, G.; Schmitt, B.; Guy, L.M.; Germond, J.-E.; Zuber, S.; Michot, L.; Reuteler, G.; Brüssow, H. Amplification and Purification of T4-Like Escherichia coli Phages for Phage Therapy: From Laboratory to Pilot Scale. Appl. Environ. Microbiol. 2014, 80, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Middelboe, M. Bacterial Growth Rate and Marine Virus–Host Dynamics. Microb. Ecol. 2000, 40, 114–124. [Google Scholar] [CrossRef]
- Nabergoj, D.; Modic, P.; Podgornik, A. Effect of bacterial growth rate on bacteriophage population growth rate. MicrobiologyOpen 2018, 7, e00558. [Google Scholar] [CrossRef] [Green Version]
- Gordillo, F.; Barr, J.J. Screening for Lysogen Activity in Therapeutically Relevant Bacteriophages. Bio-Protocol 2021, 11, e3997. [Google Scholar] [CrossRef] [PubMed]
- Merril, C.R.; Scholl, D.; Adhya, S.L. The prospect for bacteriophage therapy in Western medicine. Nat. Rev. Drug Discov. 2003, 2, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.P.; Boas, D.V.; Sillankorva, S.; Azeredo, J. Phage Therapy: A Step Forward in the Treatment of Pseudomonas aeruginosa Infections. J. Virol. 2015, 89, 7449–7456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmelcher, M.; Loessner, M.J. Bacteriophage endolysins—extending their application to tissues and the bloodstream. Curr. Opin. Biotechnol. 2021, 68, 51–59. [Google Scholar] [CrossRef]
- Ward, J.; Branston, S.; Stanley, E.; Keshavarz-Moore, E. Bacteriophages—Perspectives and Future. In Scale-Up and Bioprocessing of Phages; Savva, R., Ed.; IntechOpen: Rijeka, Croatia, 2020. [Google Scholar] [CrossRef] [Green Version]
- Schmerer, M.; Molineux, I.J.; Bull, J.J. Synergy as a rationale for phage therapy using phage cocktails. PeerJ 2014, 2, e590. [Google Scholar] [CrossRef] [Green Version]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J.; et al. Personalized Therapeutic Cocktail of Wild Environmental Phages Rescues Mice from Acinetobacter baumannii Wound Infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef] [Green Version]
- Moller, A.G.; Winston, K.; Ji, S.; Wang, J.; Hargita Davis, M.N.; Solis-Lemus, C.R.; Read, T. Genes influencing phage host range in Staphylococcus aureus on a species-wide scale. bioRxiv 2020. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Years | Authors and Study | Results and Outcome |
|---|---|---|
| 2006 | Raya et al. studied:
|
|
| 2008 | Niu et al.:
|
|
| 2011 | Vandersteegen et al. described studies on the Staphylococcus aureus phage infection parameter in two separate papers:
|
|
| 2011 | Cooper et al. studied P. aeruginosa phages’ efficacy with:
|
|
| 2013 | Henry et al.:
|
|
| 2014 | Wong et al.
|
|
| 2017 | Green et al. performed:
|
|
| 2013–2019 |
|
|
| 2018 | Xie et al. measured phage host range and “virulence” for 15 Salmonella phages using:
|
|
| 2018 | Forti et al. tested a six-phage cocktail against P. aeruginosa, which had been designed based on host range and genomic information:
|
|
| 2020 | Storms et al. and Konopacki et al., respectively [13,78]:
|
|
| 2021 | Nale et al.:
| The phage cocktail showed:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glonti, T.; Pirnay, J.-P. In Vitro Techniques and Measurements of Phage Characteristics That Are Important for Phage Therapy Success. Viruses 2022, 14, 1490. https://doi.org/10.3390/v14071490
Glonti T, Pirnay J-P. In Vitro Techniques and Measurements of Phage Characteristics That Are Important for Phage Therapy Success. Viruses. 2022; 14(7):1490. https://doi.org/10.3390/v14071490
Chicago/Turabian StyleGlonti, Tea, and Jean-Paul Pirnay. 2022. "In Vitro Techniques and Measurements of Phage Characteristics That Are Important for Phage Therapy Success" Viruses 14, no. 7: 1490. https://doi.org/10.3390/v14071490