
Global Distribution and Natural Recombination of Hepatitis D Virus: Implication of Kyrgyzstan Emerging HDVs in the Clinical Outcomes


Abstract
:1. Background
2. Methods
2.1. Phylogenetic Analysis of HDV
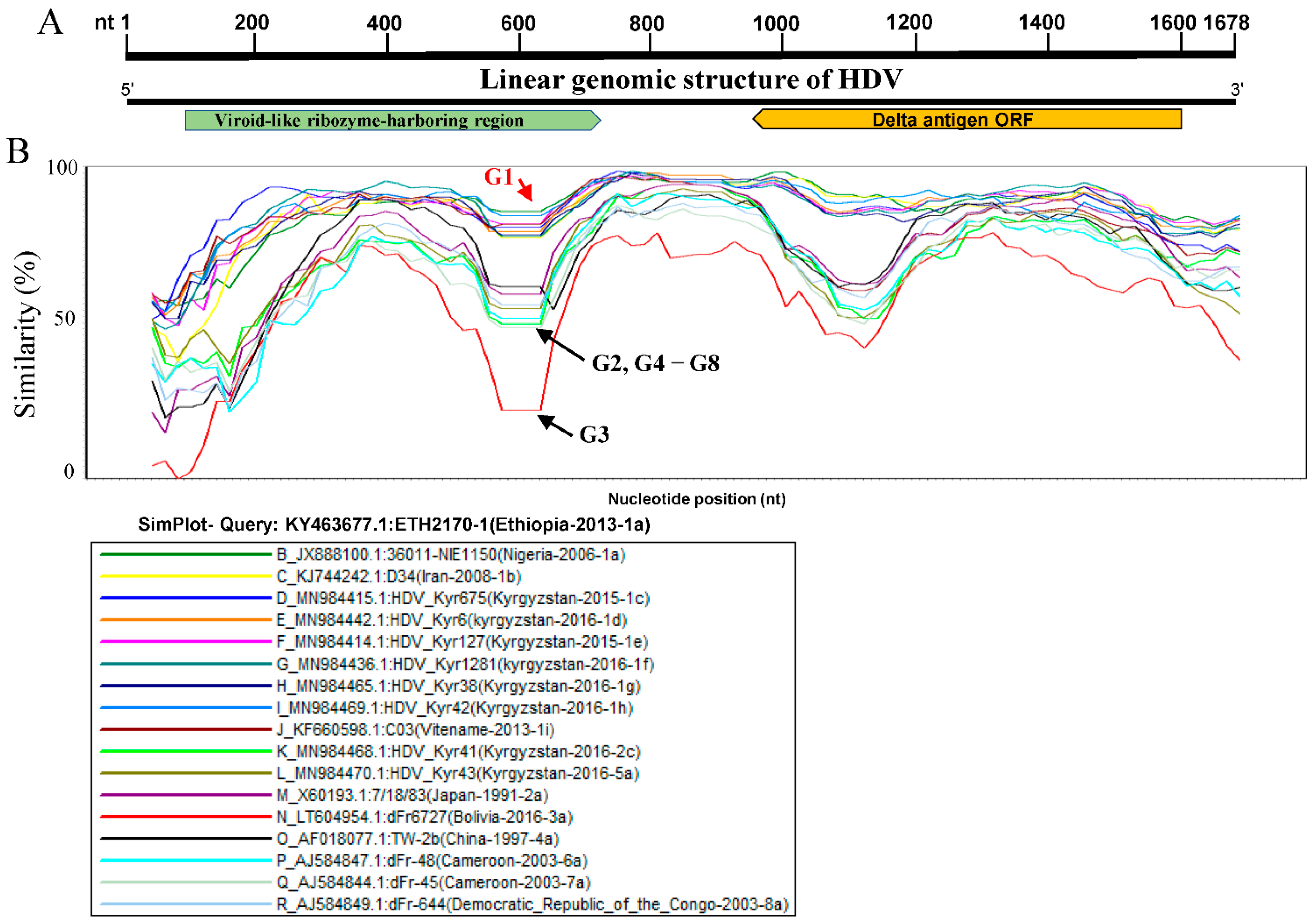
2.2. Similarity Analysis
2.3. HDV RNA Recombination
3. Results
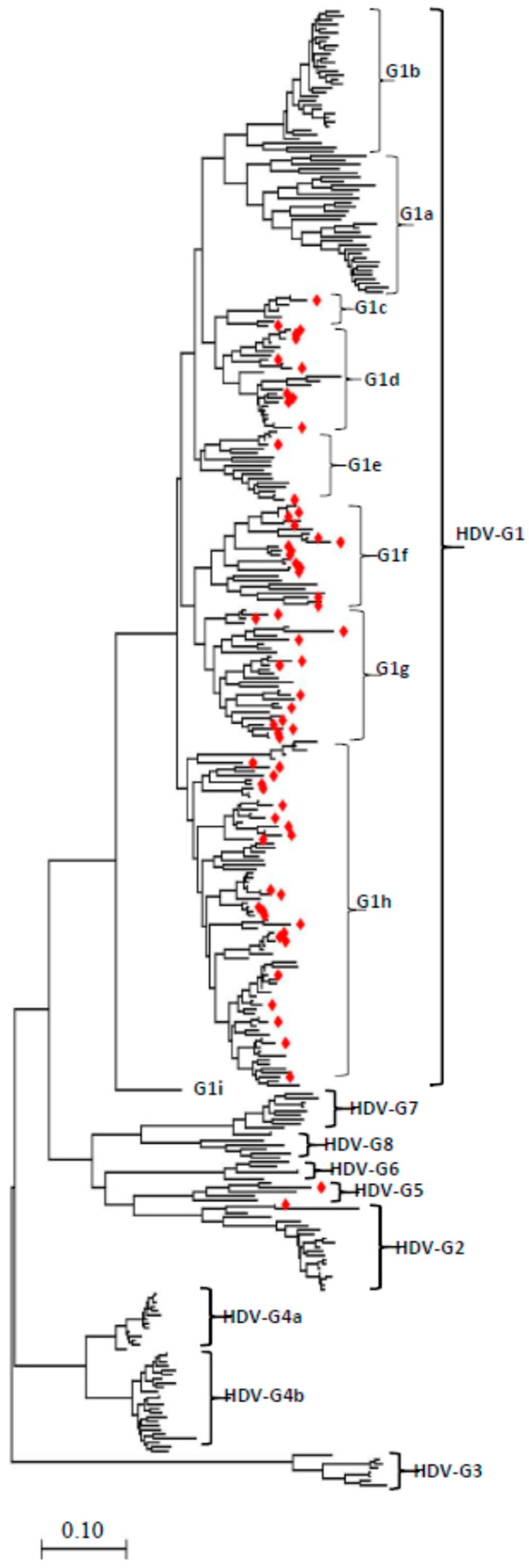
3.1. Phylogenetic Analysis of the Full-Length HDV Genome Sequences
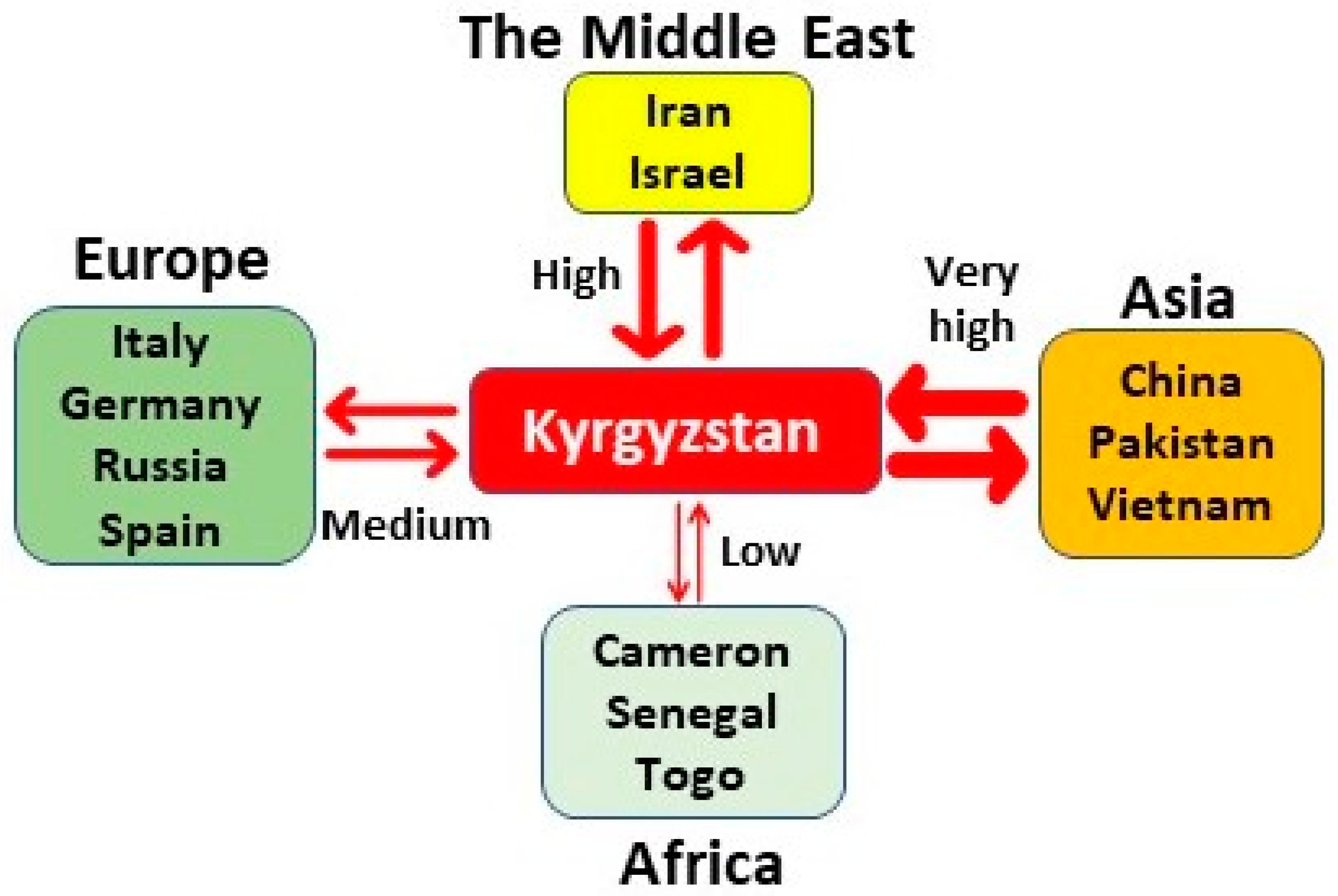
3.2. Kyrgyzstan in the Worldwide HDV Genotype Distribution
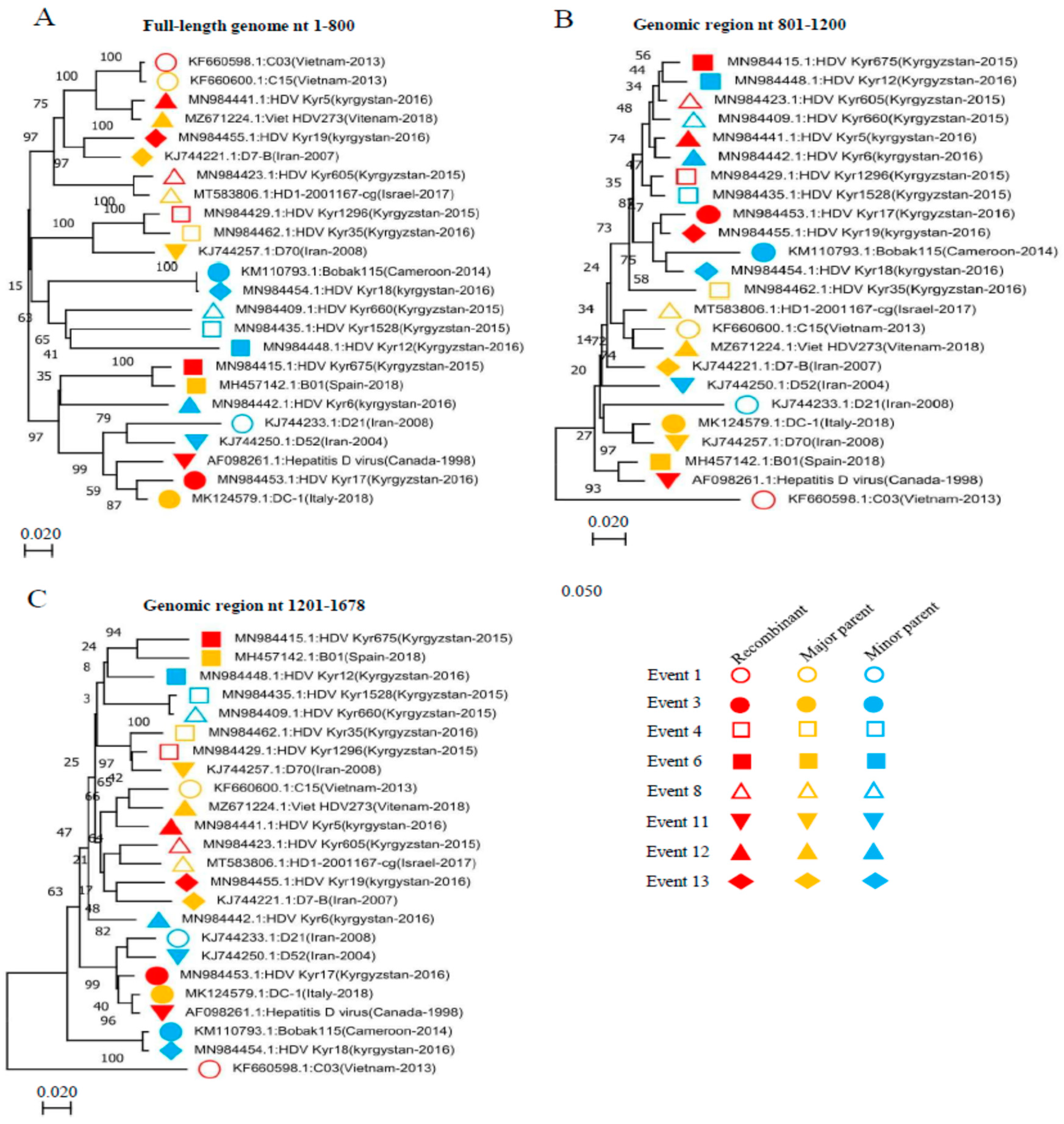
3.3. HDV Inter-Genotype Naturally Occurring Recombinant
3.4. HDV Inter-Sub-Genotype Naturally Occurring Recombinants
3.5. Identification of the Breakpoints for the HDV Recombinants
3.6. Verification of the Identified HDV Recombination Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Manka, P.; Verheyen, J.; Gerken, G.; Canbay, A. Liver Failure due to Acute Viral Hepatitis (A–E). Visc. Med. 2016, 32, 80–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzetto, M.; Hoyer, B.; Canese, M.G.; Shih, J.W.; Purcell, R.H.; Gerin, J.L. delta Agent: Association of delta antigen with hepatitis B surface antigen and RNA in serum of delta-infected chimpanzees. Proc. Natl. Acad. Sci. USA 1980, 77, 6124–6128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, H.; Siederdissen, C.; Cornberg, M. Management of HBV and HBV/HDV-Associated Liver Cirrhosis. Visc. Med. 2016, 32, 86–94. [Google Scholar] [CrossRef] [Green Version]
- WHO. Hepatitis D. 28 July 2021. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-d (accessed on 3 March 2022).
- Stockdale, A.J.; Kreuels, B.; Henrion, M.Y.R.; Giorgi, E.; Kyomuhangi, I.; de Martel, C.; Hutin, Y.; Geretti, A.M. The global prevalence of hepatitis D virus infection: Systematic review and meta-analysis. J. Hepatol. 2020, 73, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2017, 46, D708–D717. [Google Scholar] [CrossRef] [Green Version]
- Rizzetto, M.; Canese, M.G.; Arico, S.; Crivelli, O.; Trepo, C.; Bonino, F.; Verme, G. Immunofluorescence detection of new antigen-antibody system (delta/anti-delta) associated to hepatitis B virus in liver and in serum of HBsAg carriers. Gut 1977, 18, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- William Tong, C.Y.; Asher, R.; Toby, M.; Ngui, S.L.; Tettmar, K.; Ijaz, S.; Tedder, R.; Kulasegaram, R.; Wilkinson, M.; Wong, T. A re-assessment of the epidemiology and patient characteristics of hepatitis D virus infection in inner city London. J. Infect. 2013, 66, 521–527. [Google Scholar] [CrossRef]
- Heidrich, B.; Deterding, K.; Tillmann, H.L.; Raupach, R.; Manns, M.P.; Wedemeyer, H. Virological and clinical characteristics of delta hepatitis in Central Europe. J. Viral Hepat. 2009, 16, 883–894. [Google Scholar] [CrossRef]
- Amini, N.; Alavian, S.M.; Kabir, A.; Aalaei-Andabili, S.H.; Saiedi Hosseini, S.Y.; Rizzetto, M. Prevalence of hepatitis d in the eastern mediterranean region: Systematic review and meta analysis. Hepat. Mon. 2013, 13, e8210. [Google Scholar] [CrossRef] [Green Version]
- Braga, W.S.; Castilho Mda, C.; Borges, F.G.; Leão, J.R.; Martinho, A.C.; Rodrigues, I.S.; Azevedo, E.P.; Júnior, G.M.B.; Paraná, R. Hepatitis D virus infection in the Western Brazilian Amazon—Far from a vanishing disease. Rev. Da Soc. Bras. Med. Trop. 2012, 45, 691–695. [Google Scholar] [CrossRef] [Green Version]
- Stockdale, A.J.; Chaponda, M.; Beloukas, A.; Phillips, R.O.; Matthews, P.C.; Papadimitropoulos, A.; King, S.; Bonnett, L.; Geretti, A.M. Prevalence of hepatitis D virus infection in sub-Saharan Africa: A systematic review and meta-analysis. Lancet Glob. Health 2017, 5, e992–e1003. [Google Scholar] [CrossRef] [Green Version]
- Mese, S.; Nergiz, S.; Tekes, S.; Gul, K. Seroprevalence of serum HBsAg positivity and hepatitis delta virus infection among blood donors in Southeastern Turkey. Clin. Ter. 2014, 165, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Tahaei, S.M.; Mohebbi, S.R.; Azimzadeh, P.; Behelgardi, A.; Sanati, A.; Mohammadi, P.; Khanyaghma, M.; Hosseini Razavi, A.; Sharifian, A.; Zali, M.R. Prevalence of hepatitis D virus in hepatitis B virus infected patients referred to Taleghani hospital, Tehran, Iran. Gastroenterol. Hepatol. Bed Bench 2014, 7, 144–150. [Google Scholar] [PubMed]
- Jackson, K.; Tekoaua, R.; Holgate, T.; Edwards, R.; Yuen, L.; Lee, A.; Nicholson, S.; Littlejohn, M.; Locarnini, S.; Tuneti, K. Hepatitis B and D in the Pacific Islands of Kiribati. J. Clin. Virol. 2020, 129, 104527. [Google Scholar] [CrossRef]
- Magnius, L.; Taylor, J.; Mason, W.S.; Sureau, C.; Dény, P.; Norder, H.; Ictv Report, C. Virus Taxonomy Profile: Deltavirus. J. Gen. Virol. 2018, 99, 1565–1566. [Google Scholar] [CrossRef]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef]
- Sureau, C.; Moriarty, A.M.; Thornton, G.B.; Lanford, R.E. Production of infectious hepatitis delta virus in vitro and neutralization with antibodies directed against hepatitis B virus pre-S antigens. J. Virol. 1992, 66, 1241–1245. [Google Scholar] [CrossRef] [Green Version]
- Flores, R.; Owens, R.A.; Taylor, J. Pathogenesis by subviral agents: Viroids and hepatitis delta virus. Curr. Opin. Virol. 2016, 17, 87–94. [Google Scholar] [CrossRef]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, J.M. Replication of the hepatitis delta virus RNA genome. Adv. Virus Res. 2009, 74, 103–121. [Google Scholar]
- Chang, J.; Nie, X.; Chang, H.E.; Han, Z.; Taylor, J. Transcription of hepatitis delta virus RNA by RNA polymerase II. J. Virol. 2008, 82, 1118–1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, C.H.T.; Lupták, A. HDV-like self-cleaving ribozymes. RNA Biol. 2011, 8, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureau, C.; Negro, F. The hepatitis delta virus: Replication and pathogenesis. J. Hepatol. 2016, 64, S102–S116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonino, F.; Heermann, K.H.; Rizzetto, M.; Gerlich, W.H. Hepatitis delta virus: Protein composition of delta antigen and its hepatitis B virus-derived envelope. J. Virol. 1986, 58, 945–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, A.J.; Choo, Q.L.; Wang, K.S.; Govindarajan, S.; Redeker, A.G.; Gerin, J.L.; Houghton, M. A single antigenomic open reading frame of the hepatitis delta virus encodes the epitope(s) of both hepatitis delta antigen polypeptides p24 delta and p27 delta. J. Virol. 1988, 62, 594–599. [Google Scholar] [CrossRef] [Green Version]
- King, A.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. (Eds.) Genus—Deltavirus. In Virus Taxonomy; Elsevier: San Diego, CA, USA, 2012; pp. 763–766. [Google Scholar]
- Luo, G.X.; Chao, M.; Hsieh, S.Y.; Sureau, C.; Nishikura, K.; Taylor, J. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990, 64, 1021–1027. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA polymerase II elongation by hepatitis delta antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef] [Green Version]
- Deny, P. Hepatitis delta virus genetic variability: From genotypes I, II, III to eight major clades? Curr. Top Microbiol. Immunol. 2006, 307, 151–171. [Google Scholar] [CrossRef]
- Le Gal, F.; Brichler, S.; Drugan, T.; Alloui, C.; Roulot, D.; Pawlotsky, J.M.; Dény, P.; Gordien, E. Genetic diversity and worldwide distribution of the deltavirus genus: A study of 2152 clinical strains. Hepatology 2017, 66, 1826–1841. [Google Scholar] [CrossRef] [Green Version]
- Le Gal, F.; Gault, E.; Ripault, M.P.; Serpaggi, J.; Trinchet, J.C.; Gordien, E.; Dény, P. Eighth major clade for hepatitis delta virus. Emerg. Infect. Dis. 2006, 12, 1447–1450. [Google Scholar] [CrossRef] [PubMed]
- Miao, Z.; Zhang, S.; Ma, Z.; Hakim, M.S.; Wang, W.; Peppelenbosch, M.P.; Pan, Q. Recombinant identification, molecular classification and proposed reference genomes for hepatitis delta virus. J. Viral Hepat. 2019, 26, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira-Lima, F.S.; Botelho-Souza, L.F.; Roca, T.P.; Santos, A.O.d.; Oliveira, S.d.C.; Queiroz, J.A.d.S.; Santos-Alves, F.A.G.d.; Salcedo, J.M.V.; Vieira, D.S. Phylodynamic and phylogeographic analysis of hepatitis delta virus genotype 3 isolated in South America. Viruses 2019, 11, 995. [Google Scholar] [CrossRef] [Green Version]
- Su, C.W.; Huang, Y.H.; Huo, T.I.; Shih, H.H.; Sheen, I.J.; Chen, S.W.; Lee, P.C.; Lee, S.D.; Wu, J.C. Genotypes and viremia of hepatitis B and D viruses are associated with outcomes of chronic hepatitis D patients. Gastroenterology 2006, 130, 1625–1635. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.C. Functional and clinical significance of hepatitis D virus genotype II infection. Curr. Top Microbiol. Immunol. 2006, 307, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Wranke, A.; Pinheiro Borzacov, L.M.; Parana, R.; Lobato, C.; Hamid, S.; Ceausu, E.; Dalekos, G.N.; Rizzetto, M.; Turcanu, A.; Niro, G.A.; et al. Clinical and virological heterogeneity of hepatitis delta in different regions world-wide: The Hepatitis Delta International Network (HDIN). Liver Int. 2018, 38, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lempp, F.A.; Schlund, F.; Walter, L.; Decker, C.C.; Zhang, Z.; Ni, Y.; Urban, S. Assembly and infection efficacy of hepatitis B virus surface protein exchanges in 8 hepatitis D virus genotype isolates. J. Hepatol. 2021, 75, 311–323. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-C.; Lee, C.-C.; Lin, S.-H.; Huang, P.-J.; Li, H.-P.; Chang, Y.-S.; Tang, P.; Chao, M. RNA recombination in Hepatitis delta virus: Identification of a novel naturally occurring recombinant. J. Microbiol. Immunol. Infect. 2017, 50, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Microbiol. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Sy, B.T.; Nguyen, H.M.; Toan, N.L.; Song, L.H.; Tong, H.V.; Wolboldt, C.; Binh, V.Q.; Kremsner, P.G.; Velavan, T.P.; Bock, C.T. Identification of a natural intergenotypic recombinant hepatitis delta virus genotype 1 and 2 in Vietnamese HBsAg-positive patients. J. Viral Hepat. 2015, 22, 55–63. [Google Scholar] [CrossRef]
- Razavi-Shearer, D.; Gamkrelidze, I.; Nguyen, M.H.; Chen, D.S.; Van Damme, P.; Abbas, Z.; Abdulla, M.; Abou Rached, A.; Adda, D.; Aho, I.; et al. Global prevalence, treatment, and prevention of hepatitis B virus infection in 2016: A modelling study. Lancet Gastroenterol. Hepatol. 2018, 3, 383–403. [Google Scholar] [CrossRef]
- WHO. Assessment of the Viral Hepatitis Response in Kyrgyzstan 11–15 July 2016; World Health Orgzanization Regional Office for Europe: Geneva, Switzerland, 2016.
- Semenov, A.V.; Ostankova, Y.V.; Nogoybaeva, K.A.; Kasymbekova, K.T.; Lavrentieva, I.N.; Tobokalova, S.T.; Totolian, A.A. Molecular epidemiology features of HBV/HDV co-infection in Kyrgyzstan. Russ. J. Infect. Immun. 2016, 6, 141–150. [Google Scholar] [CrossRef] [Green Version]
- World Food Programme (WFP); International Organization for Migration (IOM). Migration Food Security and Nutrition in the Kyrgyz Republic; IOM: Bishkek, Kyrgyz Republic, 2021; p. 17. [Google Scholar]
- Kiesslich, D.; Crispim, M.A.; Santos, C.; Ferreira Fde, L.; Fraiji, N.A.; Komninakis, S.V.; Diaz, R.S. Influence of hepatitis B virus (HBV) genotype on the clinical course of disease in patients coinfected with HBV and hepatitis delta virus. J. Infect Dis. 2009, 199, 1608–1611. [Google Scholar] [CrossRef] [Green Version]
- Sagnelli, C.; Pisaturo, M.; Curatolo, C.; Codella, A.V.; Coppola, N.; Sagnelli, E. Hepatitis B virus/hepatitis D virus epidemiology: Changes over time and possible future influence of the SARS-CoV-2 pandemic. World J. Gastroenterol. 2021, 27, 7271–7284. [Google Scholar] [CrossRef]
- Spaan, M.; Carey, I.; Bruce, M.; Shang, D.; Horner, M.; Dusheiko, G.; Agarwal, K. Hepatitis delta genotype 5 is associated with favourable disease outcome and better response to treatment compared to genotype 1. J. Hepatol. 2020, 72, 1097–1104. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HDV Genotype | Africa | Middle East | Europe | Asia | America | Oceania | |
|---|---|---|---|---|---|---|---|
| G1 | a | Cameroon, Ethiopia, Nigeria, Somalia | Iran | Pakistan | |||
| b | Iran | Germany | Kiribati | ||||
| c | Iran, Israel | Italy, Spain | Kyrgyzstan | USA | |||
| d | The Central African Republic | Iran | Germany, Italy, Spain | China, Kyrgyzstan | Canada, USA | ||
| e | Iran, Israel | Kyrgyzstan, Pakistan | |||||
| f | Cameroon, Ethiopia, Nigeria, | Iran, | Germany | Kyrgyzstan, Pakistan | |||
| g | Iran, Israel | Kyrgyzstan | |||||
| h | Iran, Israel, Turkey | Germany, Italy, Russia | China, Japan, Kyrgyzstan, Vietnam | Brazil, USA | |||
| i | Vietnam | ||||||
| G2 | a | China (Taiwan), Japan, Vietnam | |||||
| b | Russia | China (Taiwan) | |||||
| c | Kyrgyzstan, Vietnam | ||||||
| G3 | a | Bolivia | |||||
| b | Brazil, Venezuela | ||||||
| c | Brazil | ||||||
| G4 | a | China, China (Taiwan), Japan | |||||
| b | Japan | ||||||
| G5 | a | Nigeria, Senegal | Kyrgyzstan | ||||
| b | Guinea-Bissau, Togo | ||||||
| G6 | a | Cameroon | |||||
| b | Nigeria | ||||||
| c | The Central African Republic, | ||||||
| G7 | a | Cameroon | |||||
| b | Cameroon | ||||||
| G8 | a | Democratic Republic of the Congo, Cote d’Ivoire, | |||||
| b | Democratic Republic of the Congo, Gabon, Senegal | ||||||
| Recombination Event Serial Number | Recombinant | Minor Parent | Major Parent | Detection Methods | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GenBank ID: Virus Name (Country-Year) | Genotype | GenBank ID: Virus Name (Country-Year) | Genotype | GenBank ID: Virus Name (Country-Year) | Genotype | R | G | B | M | C | S | T | |
| 1 | KF660598.1:C03 (Vietnam-2013) | G1i | KJ744233.1:D21 (Iran-2008) | G1d | KF660600.1:C15 (Vietnam-2013) | G1h | + | + | + | + | + | - | + |
| 2 | * KJ744233.1:D21 (Iran-2008) | G1d | HM046802.1:JN (China-2010) | G1d | KJ744257.1:D70 (Iran-2008) | G1g | + | + | + | + | - | + | + |
| 3 | MN984453.1:HDV_Kyr17 (Kyrgyzstan-2016) | G1d | KM110793.1:Bobak115 (Cameroon-2014) | G1f | MK124579.1:DC-1 (Italy-2018) | G1d | + | + | + | + | + | + | + |
| 4 | * MN984429.1:HDV_Kyr1296 (Kyrgyzstan-2015) | G1g | MN984435.1:HDV_Kyr1528 (Kyrgyzstan-2015) | G1f | MN984462.1:HDV_Kyr35 (Kyrgyzstan-2016) | G1g | + | + | + | + | + | + | + |
| 5 | MN984449.1:HDV_Kyr13 (Kyrgyzstan-2016) | G1d | MN984446.1:HDV_Kyr10 (Kyrgyzstan-2016) | G1g | HM046802.1:JN (China-2010) | G1d | + | + | + | + | + | + | + |
| 6 | MN984415.1:HDV_Kyr675 (Kyrgyzstan-2015) | G1c | MN984448.1:HDV_Kyr12 (kygyzstan-2016) | G1f | MH457142.1:B01 (Spain-2018) | G1c | + | + | + | + | + | + | + |
| 7 | MN984432.1:HDV_Kyr857 (Kyrgyzstan-2015) | G1h | MN984448.1:HDV_Kyr12 (kygyzstan-2016) | G1f | KJ744221.1:D7-B (Iran-2007) | G1h | + | + | + | + | + | + | + |
| 8 | MN984423.1:HDV_Kyr605 (Kyrgyzstan-2015) | G1h | MN984409.1:HDV_Kyr660 (Kyrgyzstan-2015) | G1f | MT583806.1:HD1-2001167-cg (Israel-2017) | G1h | + | + | + | + | + | + | + |
| 9 | * MN984435.1:HDV_Kyr1528 (Kyrgyzstan-2015) | G1f | MN984436.1:HDV_Kyr1281 (Kyrgyzstan-2016) | G1f | MN984416.1:HDV_Kyr393 (Kyrgyzstan-2015) | G1f | + | + | + | + | + | + | + |
| 10 | MN984414.1:HDV_Kyr127 (Kyrgyzstan-2015) | G1e | MN984448.1:HDV_Kyr12 (Kygyzstan-2016) | G1f | KJ744215.1:D2-A (Iran-2006) | G1e | + | + | - | + | + | + | + |
| 11 | AF098261.1:Hepatitis_D_virus (Canada-1998) | G1d | KJ744250.1:D52 (Iran-2004) | G1d | KJ744257.1:D70 (Iran-2008) | G1g | + | + | + | + | + | + | + |
| 12 | MN984441.1:HDV_Kyr5 (Kyrgyzstan-2016) | G1h | MN984442.1:HDV_Kyr6 (Kyrgyzstan-2016) | G1d | MZ671224.1:Viet_HDV273 (Vietnam-2018) | G1h | + | + | + | + | + | + | + |
| 13 | * MN984455.1:HDV_Kyr19 (Kyrgyzstan-2016) | G1h | MN984454.1:HDV_Kyr18 (Kyrgyzstan-2016) | G1f | KJ744221.1:D7-B (Iran-2007) | G1h | + | + | + | + | + | + | + |
| 14 | * KJ744253.1:D60 (Iran-2007) | G1f | MN984448.1:HDV_Kyr12 (Kygyzstan-2016) | G1f | MN984409.1:HDV_Kyr660 (Kyrgyzstan-2015) | G1f | + | + | + | + | + | + | + |
| 15 | * KJ744250.1:D52 (Iran-2004) | G1d | AF098261.1:Hepatitis_D_virus (Canada-1998) | G1d | KJ744230.1:D15 (Iran-2004) | G1c | + | + | + | + | + | + | + |
| 16 | KM110792.1:Bobak66 (Cameroon-2014) | G1a | MN984427.1:HDV_Kyr893 (Kyrgyzstan-2015) | G1f | KM110794.1:Bobak118 (Cameroon-2014) | G1a | + | - | + | + | + | - | + |
| 17 | * MN984454.1:HDV_Kyr18 (Kyrgyzstan-2016) | G1f | MN984412.1:HDV_Kyr599 (Kyrgyzstan-2015) | G1f | KM110793.1:Bobak115 (Cameroon-2014) | G1f | + | + | - | - | - | + | + |
| 18 | MN984438.1:HDV_Kyr2 (Kyrgyzstan-2016) | G1h | MN984442.1:HDV_Kyr6 (Kyrgyzstan-2016) | G1d | AY648957.1:TW5132*24 (Taiwan-2004) | G1h | + | + | + | + | + | + | + |
| 19 | * MN984418.1:HDV_Kyr476 (Kyrgyzstan-2015) | G1g | MN984440.1:HDV_Kyr4 (Kyrgyzstan-2016) | G1h | KJ744255.1:D66 (Iran-2003) | G1g | + | + | - | + | - | + | + |
| 20 | MN984436.1:HDV_Kyr1281 (Kyrgyzstan-2016) | G1f | MN984459.1:HDV_Kyr31 (Kyrgyzstan-2016) | G1d | MN984409.1:HDV_Kyr660 (Kyrgyzstan-2015) | G1f | + | + | + | + | + | + | + |
| 21 | * MN984424.1:HDV_Kyr1279 (Kyrgyzstan-2015) | G1d | MN984448.1:HDV_Kyr12 (Kygyzstan-2016) | G1f | MH457147.1:E36-28269 (Germany-2018) | G1d | + | + | + | + | + | - | + |
| 22 | * KJ744214.1:D1 (Iran-2008) | G1f | MN984440.1:HDV_Kyr4 (Kyrgyzstan-2016) | G1h | MN984409.1:HDV_Kyr660 (Kyrgyzstan-2015) | G1f | + | + | - | - | - | - | + |
| 23 | * AM183328.1:DFr2703 (Senegal-2006) | G5a | MN984470.1:HDV_Kyr43 (Kyrgyzstan-2016) | G5a | AM183326.1:DFr2600 (Togo-2006) | G5b | + | + | + | + | + | + | + |
| 24 | * MK890231.1:QD-18 (Pakistan-2018) | G1f | MN984413.1:HDV_Kyr914 (Kyrgyzstan-2015) | G1g | MN984447.1:HDV_Kyr11 (Kyrgyzstan-2016) | G1f | + | + | - | + | + | - | + |
| 25 | * KJ744253.1:D60 (Iran-2007) | G1f | KJ744247.1:D43 (Iran-2007) | G1g | KJ744214.1:D1 (Iran-2008) | G1f | + | + | + | + | + | + | + |
| 26 | AB118845.1:Miyako (JA-M36) (Japan-2003) | G4a | AB118826.1:Miyako (JA-M11) (Japan-2003) | G4b | AY648952.1:TWD62*16 (Taiwan-2004) | G4a | + | + | - | + | + | + | + |
| 27 | * EF514906.1:SO (Turkey-2007) | G1h | MH457147.1:E36-28269 (Germany-2018) | G1d | EF514904.1:NK (Turkey-2007) | G1h | + | + | + | + | + | + | + |
| 28 | * MN984460.1:HDV_Kyr33 (Kyrgyzstan-2016) | G1h | MH791030.1:63 (Russia-2017) | G1h | MN984423.1:HDV_Kyr605 (Kyrgyzstan-2015) | G1h | + | + | + | + | + | + | + |
| 29 | * MN984456.1:HDV_Kyr24 (Kyrgyzstan-2016) | G1d | MN984442.1:HDV_Kyr6 (Kyrgyzstan-2016) | G1d | MH457150.1:H29-10701 (Germany-2018) | G1d | + | + | + | + | + | + | + |
| 30 | KJ744247.1:D43 (Iran-2007) | G1g | KJ744225.1:D9-A (Iran-2007) | G1g | KJ744248.1:D49 (Iran-2010) | G1g | + | + | + | + | + | - | + |
| 31 | * MZ671215.1:Viet_HDV270 (Vietnam-2018) | G1h | MH457149.1:H07-10501 (Germany-2018) | G1b | MN984434.1:HDV_Kyr1026 (Kyrgyzstan-2015) | G1h | + | + | + | + | + | + | + |
| 32 | * MN984459.1:HDV_Kyr31 (Kyrgyzstan-2016) | G1d | KJ744244.1:D37-B (Iran-2007) | G1g | MK124579.1:DC-1 (Italy-2018) | G1d | + | + | - | + | + | - | + |
| 33 | * MN984434.1:HDV_Kyr1026 (Kyrgyzstan-2015) | G1h | MN984458.1:HDV_Kyr30 (Kyrgyzstan-2016) | G1h | MZ671212.1:Viet_HDV44 (Vietnam-2018) | G1h | + | + | + | + | + | - | + |
| 34 | * MN984421.1:HDV_Kyr734 (Kyrgyzstan-2015) | G1f | KJ744216.1:D2-B (Iran-2007) | G1e | MK890225.1:QD-01 (Pakistan-2018) | G1f | + | + | - | + | + | + | + |
| 35 | * MN984465.1:HDV_Kyr38 (Kyrgyzstan-2016) | G1g | KJ744245.1:D39-A (Iran-2006) | G1g | KJ744244.1:D37-B (Iran-2007) | G1g | + | - | + | - | - | + | + |
| 36 | MN984428.1:HDV_Kyr1290 (Kyrgyzstan-2015) | G1d | MG711663.1:A176 (Cameroon-2015) | G1a | MH457147.1:E36-28269 (Germany-2018) | G1d | + | + | - | + | + | + | + |
| 37 | * MN984442.1:HDV_Kyr6 (Kyrgyzstan-2016) | G1d | MN984440.1:HDV_Kyr4 (Kyrgyzstan-2016) | G1h | MH457150.1:H29-10701 (Germany-2018) | G1d | + | + | - | + | + | + | + |
| 38 | * MN984445.1:HDV_Kyr9 (Kyrgyzstan-2016) | G1e | MN984419.1:HDV_Kyr850 (Kyrgyzstan-2015) | G1f | MK890232.1:QD-22 (Pakistan-2018) | G1e | + | + | + | + | + | + | + |
| 39 | * MN984463.1:HDV_Kyr36 (Kyrgyzstan-2016) | G1g | KJ744218.1:D5-A (Iran-2006) | G1g | KJ744243.1:D37-A (Iran-2007) | G1g | + | + | - | + | + | + | + |
| 40 | * MN984448.1:HDV_Kyr12 (Kyrgyzstan-2016) | G1f | MN984413.1:HDV_Kyr914 (Kyrgyzstan-2015) | G1g | MZ671223.1:Viet_HDV289 (Vietnam-2018) | G1h | + | + | + | + | + | + | + |
| 41 | * KJ744244.1:D37-B (Iran-2007) | G1g | AJ307077.1:W5 (Italy-2001) | G1d | KJ744243.1:D37-A (Iran-2007) | G1g | + | + | - | + | + | + | + |
| 42 | * KJ744225.1:D9-A (Iran-2007) | G1g | KJ744237.1:D27 (Iran-2008) | G1e | KJ744226.1:D9-B (Iran-2008) | G1g | + | - | - | + | + | + | + |
| 43 | * MH791030.1:63 (Russia-2017) | G1h | MG711712.1:K2000 (Cameroon-2013) | G1a | MT649284.1:17500175 (Kiribati-2017) | G1b | + | - | - | + | + | + | + |
| 44 | * KJ744242.1:D34 (Iran-2008) | G1b | KJ744238.1:D28 (Iran-2008) | G1e | KJ744240.1:D32 (Iran-2008) | G1b | + | - | - | + | + | + | + |
| 45 | * MN984407.1:HDV_Kyr1376 (Kyrgyzstan-2015) | G1f | KJ744256.1:D69 (Iran-2005) | G1g | MK890225.1:QD-01 (Pakistan-2018) | G1f | + | + | - | + | + | - | - |
| 46 | * AM183326.1:DFr2600 (Togo-2006) | G5b | MN984468.1:HDV_Kyr41 (Kyrgyzstan-2016) | G2c | MN984470.1:HDV_Kyr43 (Kyrgyzstan-2016) | G5a | + | - | + | + | + | + | + |
| 47 | * MN984465.1:HDV_Kyr38 (Kyrgyzstan-2016) | G1g | KJ744245.1:D39-A (Iran-2006) | G1g | KJ744218.1:D5-A (Iran-2006) | G1g | + | + | + | - | + | + | + |
| 48 | MN984461.1:HDV_Kyr34 (Kyrgyzstan-2016) | G1h | MT583806.1:HD1-2001167-cg (Israel-2017) | G1h | MH791030.1:63 (Russia-2017) | G1h | + | + | - | + | + | - | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bahoussi, A.N.; Wang, P.-H.; Guo, Y.-Y.; Rabbani, N.; Wu, C.; Xing, L. Global Distribution and Natural Recombination of Hepatitis D Virus: Implication of Kyrgyzstan Emerging HDVs in the Clinical Outcomes. Viruses 2022, 14, 1467. https://doi.org/10.3390/v14071467
Bahoussi AN, Wang P-H, Guo Y-Y, Rabbani N, Wu C, Xing L. Global Distribution and Natural Recombination of Hepatitis D Virus: Implication of Kyrgyzstan Emerging HDVs in the Clinical Outcomes. Viruses. 2022; 14(7):1467. https://doi.org/10.3390/v14071467
Chicago/Turabian StyleBahoussi, Amina Nawal, Pei-Hua Wang, Yan-Yan Guo, Nighat Rabbani, Changxin Wu, and Li Xing. 2022. "Global Distribution and Natural Recombination of Hepatitis D Virus: Implication of Kyrgyzstan Emerging HDVs in the Clinical Outcomes" Viruses 14, no. 7: 1467. https://doi.org/10.3390/v14071467