
Identification of Phage Receptor-Binding Protein Sequences with Hidden Markov Models and an Extreme Gradient Boosting Classifier


Abstract
:1. Introduction
2. Materials and Methods
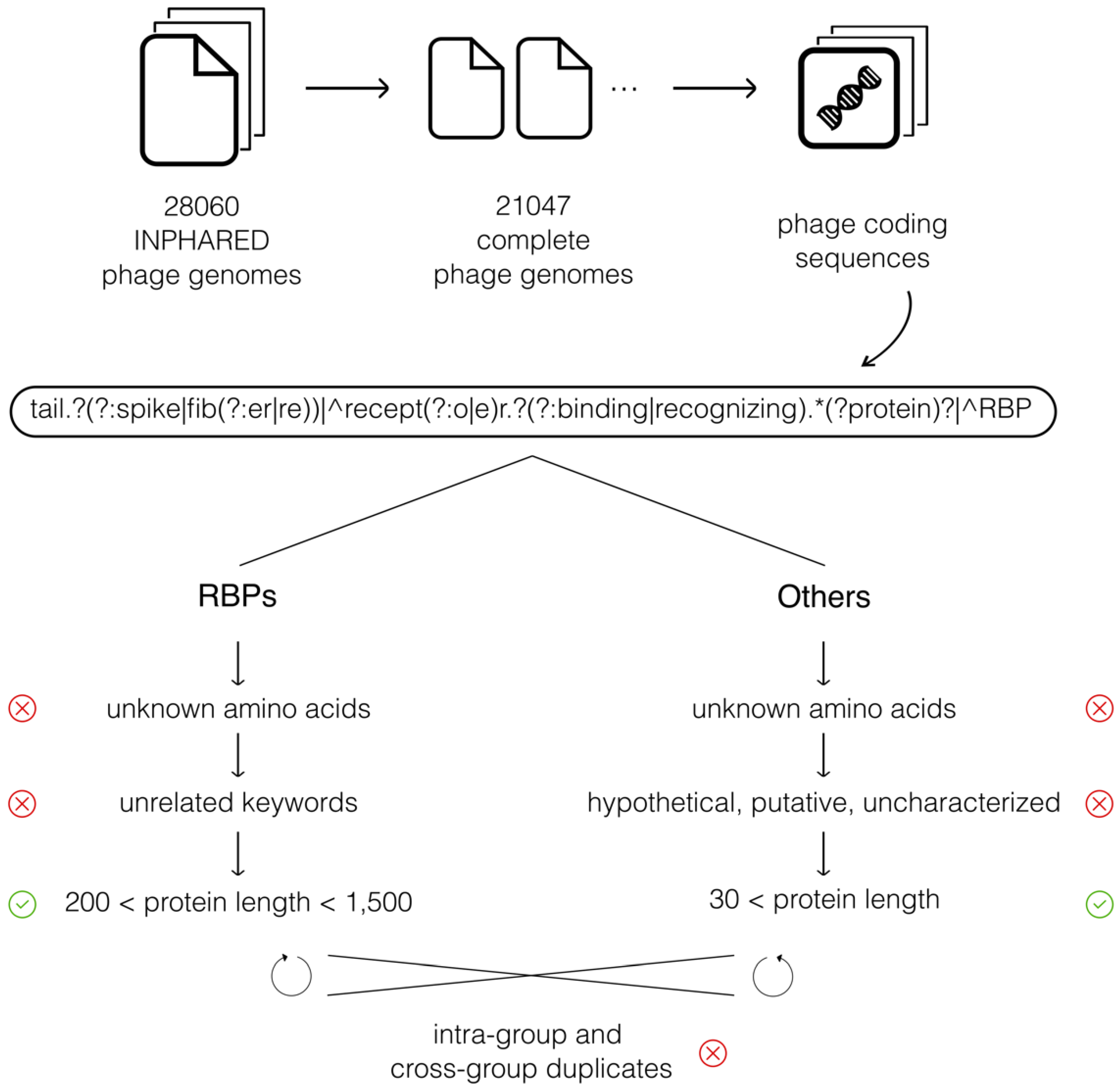
2.1. Phage Genome Sequence Data
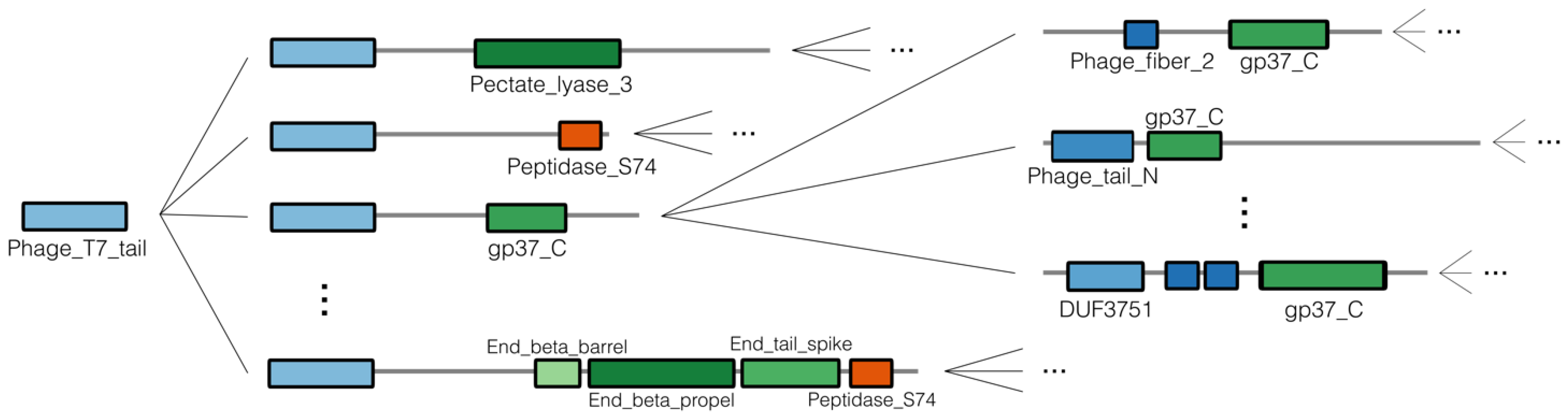
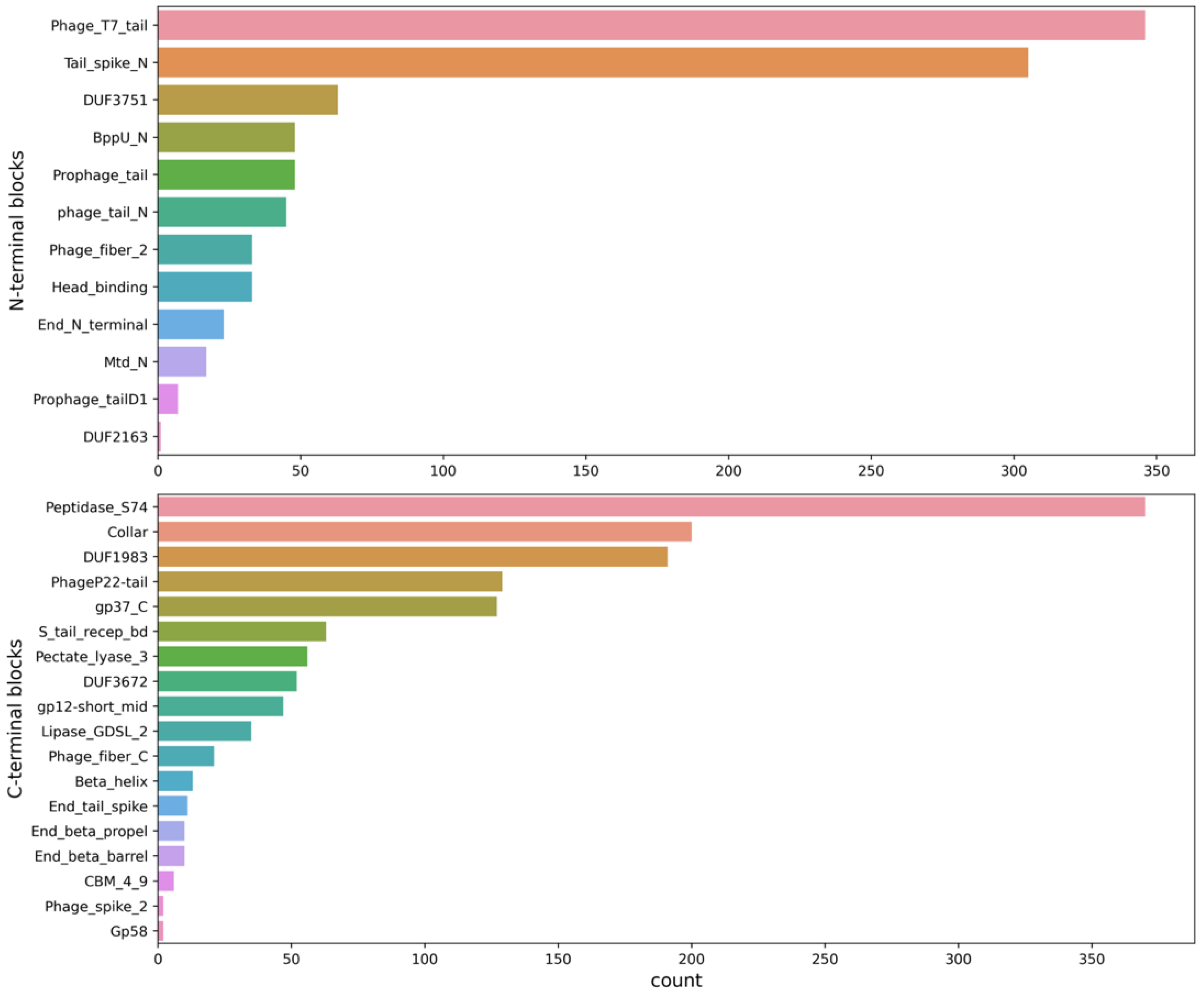
2.2. Collecting and Constructing Profile HMMs Related to RBPs
2.3. Training an Extreme Gradient Boosting Classifier to Discriminate Phage RBPs from Other Phage Proteins
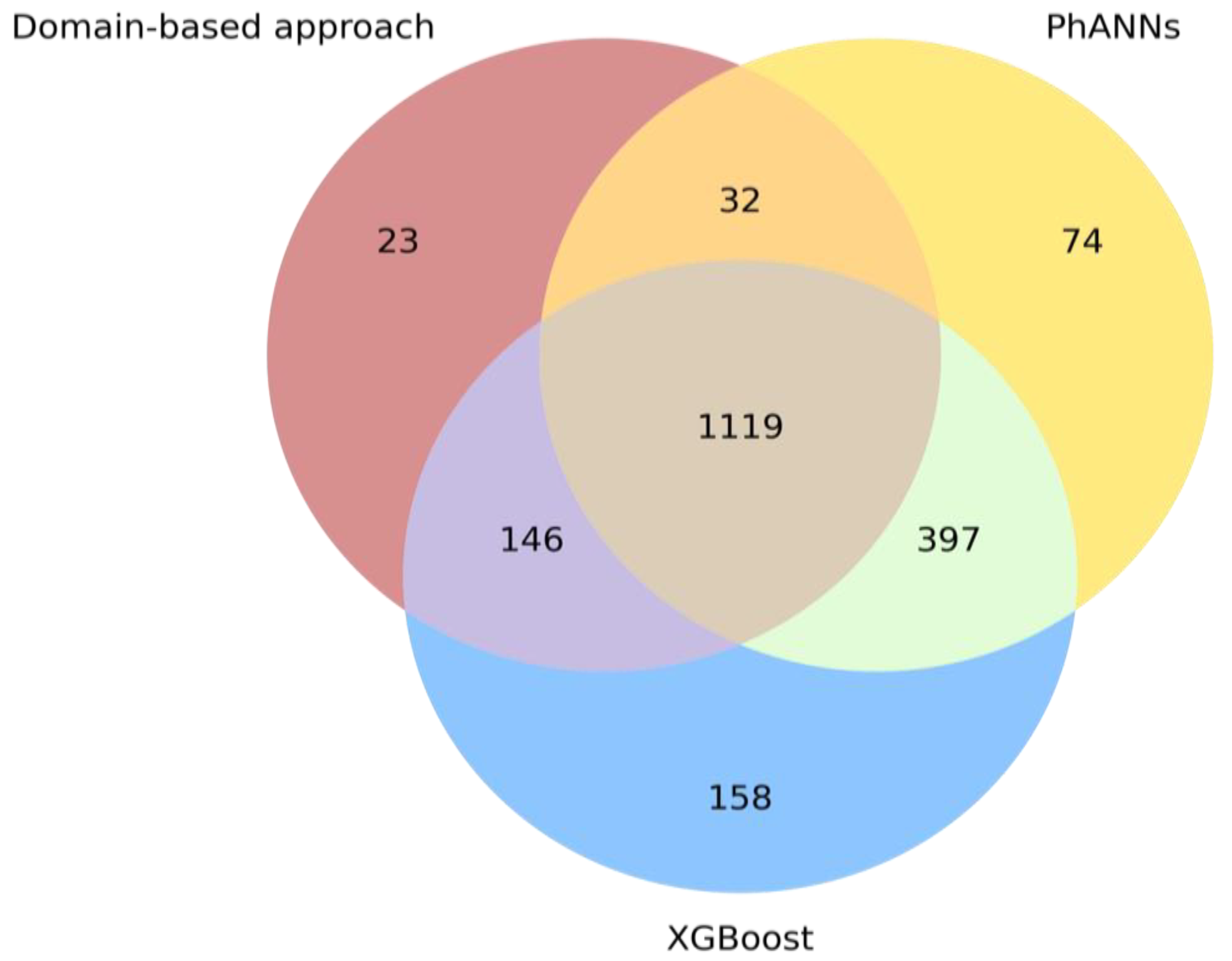
2.4. Benchmarking Our Parallel Approaches against PhANNs
3. Results
3.1. Phage Genome Sequence Data
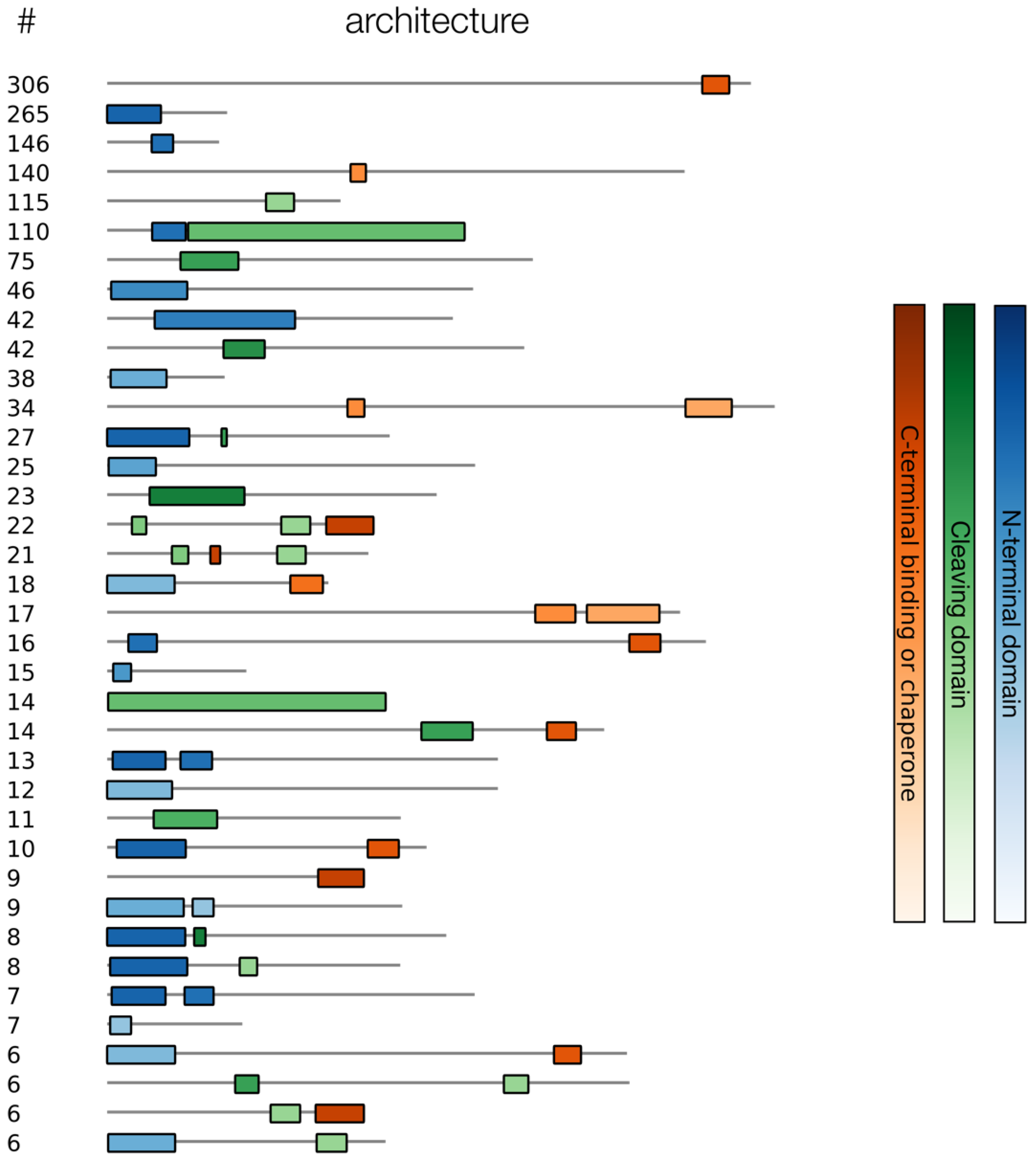
3.2. Collecting and Constructing Profile HMMs Related to RBPs
3.3. Training an Extreme Gradient Boosting Classifier to Discriminate Phage RBPs from Other Phage Proteins
3.4. Benchmarking Our Parallel Approaches against PhANNs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Kortright, K.E.; Chan, B.K.; Koff, J.L.; Turner, P.E. Phage Therapy: A Renewed Approach to Combat Antibiotic-Resistant Bacteria. Cell Host Microbe 2019, 25, 219–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eskenazi, A.; Lood, C.; Wubbolts, J.; Hites, M.; Balarjishvili, N.; Leshkasheli, L.; Askilashvili, L.; Kvachadze, L.; van Noort, V.; Wagemans, J.; et al. Combination of pre-adapted bacteriophage therapy and antibiotics for treatment of fracture-related infection due to pandrug-resistant Klebsiella pneu-moniae. Nat. Commun. 2022, 13, 302. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Meaden, S. Understanding Bacteriophage Specificity in Natural Microbial Communities. Viruses 2013, 5, 806–823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüssow, H. Hurdles for Phage Therapy to Become a Reality—An Editorial Comment. Viruses 2019, 11, 557. [Google Scholar] [CrossRef] [Green Version]
- Chan, B.K.; Abedon, S.T.; Loc-Carrillo, C. Phage cocktails and the future of phage therapy. Future Microbiol. 2013, 8, 769–783. [Google Scholar] [CrossRef]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing phage therapy through synthetic biology and genome engineering. Curr. Opin. Biotechnol. 2020, 68, 151–159. [Google Scholar] [CrossRef]
- Dunne, M.; Rupf, B.; Tala, M.; Qabrati, X.; Ernst, P.; Shen, Y.; Sumrall, E.; Heeb, L.; Plückthun, A.; Loessner, M.J.; et al. Reprogramming Bacteriophage Host Range through Structure-Guided Design of Chimeric Receptor Binding Proteins. Cell Rep. 2019, 29, 1336–1350.e4. [Google Scholar] [CrossRef] [Green Version]
- Huss, P.; Meger, A.; Leander, M.; Nishikawa, K.; Raman, S. Mapping the functional landscape of the receptor binding domain of T7 bacteriophage by deep mutational scanning. eLife 2021, 10, 63775. [Google Scholar] [CrossRef]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering Phage Host-Range and Suppressing Bacterial Resistance through Phage Tail Fiber Mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef]
- Nóbrega, F.L.; Vlot, M.; de Jonge, P.A.; Dreesens, L.L.; Beaumont, H.J.E.; Lavigne, R.; Dutilh, B.E.; Brouns, S.J.J. Targeting mechanisms of tailed bacteriophages. Nat. Rev. Microbiol. 2018, 16, 760–773. [Google Scholar] [CrossRef] [PubMed]
- Cook, R.; Brown, N.; Redgwell, T.; Rihtman, B.; Barnes, M.; Clokie, M.; Stekel, D.J.; Hobman, J.; Jones, M.A.; Millard, A. INfrastructure for a PHAge REference Database: Identification of Large-Scale Biases in the Current Collection of Cultured Phage Genomes. Phage 2021, 2, 214–223. [Google Scholar] [CrossRef]
- Lood, C.; Boeckaerts, D.; Stock, M.; De Baets, B.; Lavigne, R.; van Noort, V.; Briers, Y. Digital phagograms: Predicting phage infectivity through a multilayer machine learning approach. Curr. Opin. Virol. 2021, 52, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a fast and accurate tool and web server to classify phage structural proteins. PLoS Comput. Biol. 2020, 16, e1007845. [Google Scholar] [CrossRef]
- Yukgehnaish, K.; Rajandas, H.; Parimannan, S.; Manickam, R.; Marimuthu, K.; Petersen, B.; Clokie, M.R.J.; Millard, A.; Sicheritz-Pontén, T. PhageLeads: Rapid Assessment of Phage Therapeutic Suitability Using an Ensemble Machine Learning Approach. Viruses 2022, 14, 342. [Google Scholar] [CrossRef]
- Li, H.-F.; Wang, X.-F.; Tang, H. Predicting Bacteriophage Enzymes and Hydrolases by Using Combined Features. Front. Bioeng. Biotechnol. 2020, 8, 183. [Google Scholar] [CrossRef]
- Fernández-Ruiz, I.; Coutinho, F.H.; Rodriguez-Valera, F. Thousands of Novel Endolysins Discovered in Uncultured Phage Genomes. Front. Microbiol. 2018, 9, 1033. [Google Scholar] [CrossRef] [Green Version]
- Haggård-Ljungquist, E.; Halling, C.; Calendar, R. DNA sequences of the tail fiber genes of bacteriophage P2: Evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J. Bacteriol. 1992, 174, 1462–1477. [Google Scholar] [CrossRef] [Green Version]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef]
- Dallago, C.; Schütze, K.; Heinzinger, M.; Olenyi, T.; Littmann, M.; Lu, A.X.; Yang, K.K.; Min, S.; Yoon, S.; Morton, J.T.; et al. Learned Embeddings from Deep Learning to Visualize and Predict Protein Sets. Curr. Protoc. 2021, 1, e113. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Leiman, P.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Steven, A.; Trus, B.; Maizel, J.; Unser, M.; Parry, D.; Wall, J.; Hainfeld, J.; Studier, F. Molecular substructure of a viral receptor-recognition protein: The gp17 tail-fiber of bacteriophage T. J. Mol. Biol. 1988, 200, 351–365. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov Models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the KDD ’16: 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Elnaggar, A.; Heinzinger, M.; Dallago, C.; Rehawi, G.; Wang, Y.; Jones, L.; Gibbs, T.; Feher, T.; Angerer, C.; Steinegger, M.; et al. ProtTrans: Towards Cracking the Language of Lifes Code Through Self-Supervised Deep Learning and High Performance Computing. arXiv 2021, arXiv:2104.02443. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2012, 12, 2825–2830. [Google Scholar]
- Durbin, R.; Eddy, S.R.; Krogh, A.; Mitchison, G. Biological Sequence Analysis; Cambridge University Press: Cambridge, UK, 1998. [Google Scholar] [CrossRef]
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the T4 tail and tail fibers. Virol. J. 2010, 7, 355. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Meier, M.; Mirdita, M.; Vöhringer, H.; Haunsberger, S.J.; Söding, J. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinform. 2019, 20, 473. [Google Scholar] [CrossRef] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| |
| Pass | tailspike, tail spike, tail-fiber, receptor-binding protein |
| Fail | spike, tail protein, binding protein |
| |
| RBPs | adaptor, wedge, baseplate, hinge, connector, structural, component, assembly, chaperone, attachment, capsid, proximal, measure |
| Others | probable, probably, uncharacterized, uncharacterised, putative, hypothetical, unknown, predicted |
| N-Terminal Domain | C-Terminal Domain |
|---|---|
| Phage_T7_tail | Lipase_GDSL_2 |
| Tail_spike_N | Pectate_lyase_3 |
| Prophage_tail | gp37_C |
| BppU_N | Beta_helix |
| Mtd_N | End_beta_propel |
| Head_binding | End_tail_spike |
| DUF3751 | End_beta_barrel |
| End_N_terminal | PhageP22-tail |
| phage_tail_N | Phage_spike_2 |
| Prophage_tailD1 | gp12-short_mid |
| DUF2163 | Collar |
| Phage_fiber_2 | Peptidase_S74 |
| Phage_fiber_C | |
| S_tail_recep_bd | |
| CBM_4_9 | |
| DUF1983 | |
| DUF3672 |
| Method | F1-Score | MCC | Sensitivity | Specificity |
|---|---|---|---|---|
| Domain-based | 72.0% | 70.2% | 66.4% | 98.5% |
| PhANNs | 69.8% | 67.9% | 81.6% | 95.8% |
| XGBoost | 84.0% | 82.3% | 91.6% | 97.9% |
| XGBoost + HMM scores | 84.8% | 83.8% | 92.2% | 98.0% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boeckaerts, D.; Stock, M.; De Baets, B.; Briers, Y. Identification of Phage Receptor-Binding Protein Sequences with Hidden Markov Models and an Extreme Gradient Boosting Classifier. Viruses 2022, 14, 1329. https://doi.org/10.3390/v14061329
Boeckaerts D, Stock M, De Baets B, Briers Y. Identification of Phage Receptor-Binding Protein Sequences with Hidden Markov Models and an Extreme Gradient Boosting Classifier. Viruses. 2022; 14(6):1329. https://doi.org/10.3390/v14061329
Chicago/Turabian StyleBoeckaerts, Dimitri, Michiel Stock, Bernard De Baets, and Yves Briers. 2022. "Identification of Phage Receptor-Binding Protein Sequences with Hidden Markov Models and an Extreme Gradient Boosting Classifier" Viruses 14, no. 6: 1329. https://doi.org/10.3390/v14061329