
In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Cycloheximide Treatment
2.3. RNA Purification
2.4. Poly(A) RNA Isolation
2.5. Direct cDNA Sequencing
2.6. Pre-Processing and Data Analysis
3. Results
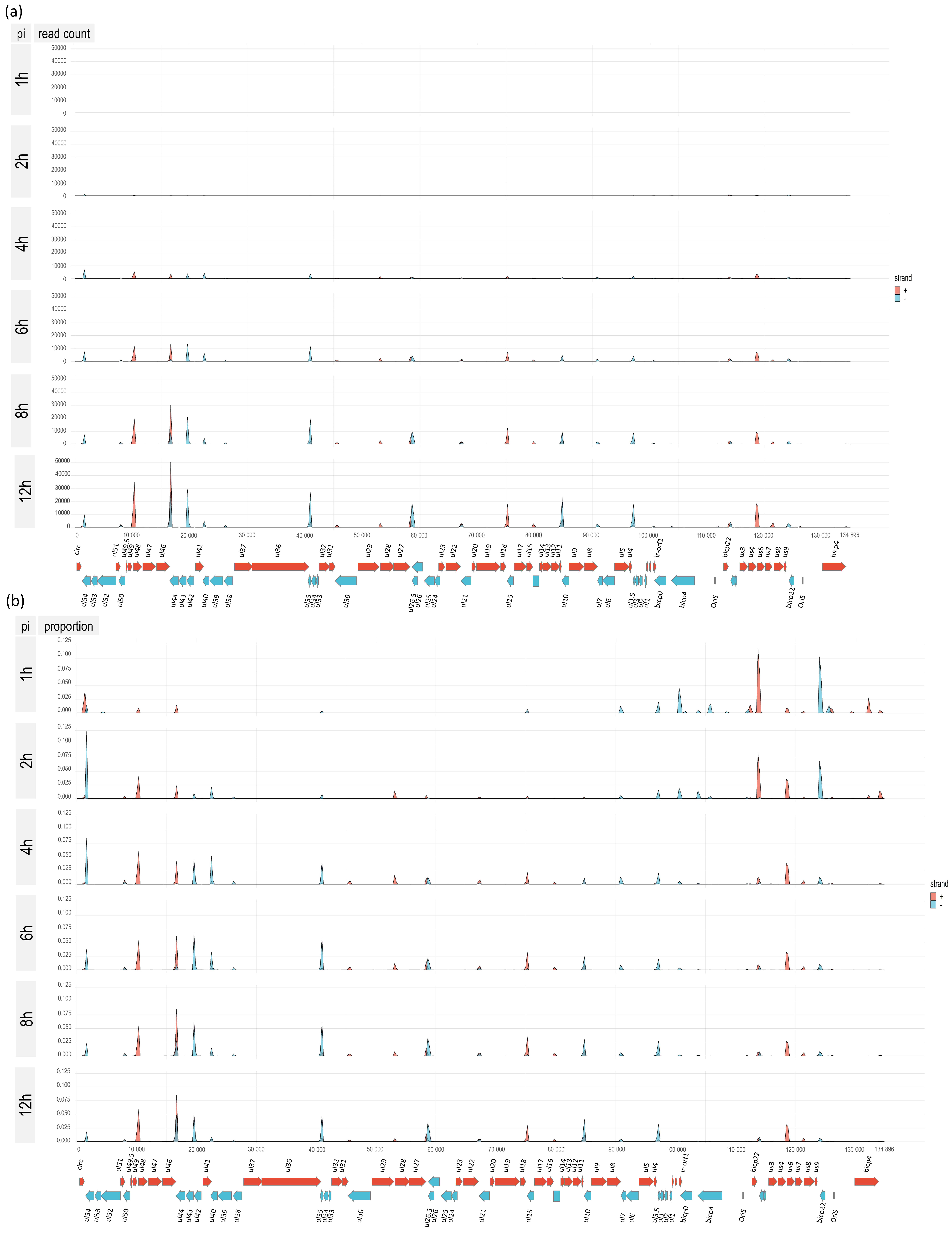
3.1. Time Course Analysis of BoHV-1 Transcriptome Using Nanopore Sequencing
3.2. BoHV-1 Expresses Four Immediate-Early Genes
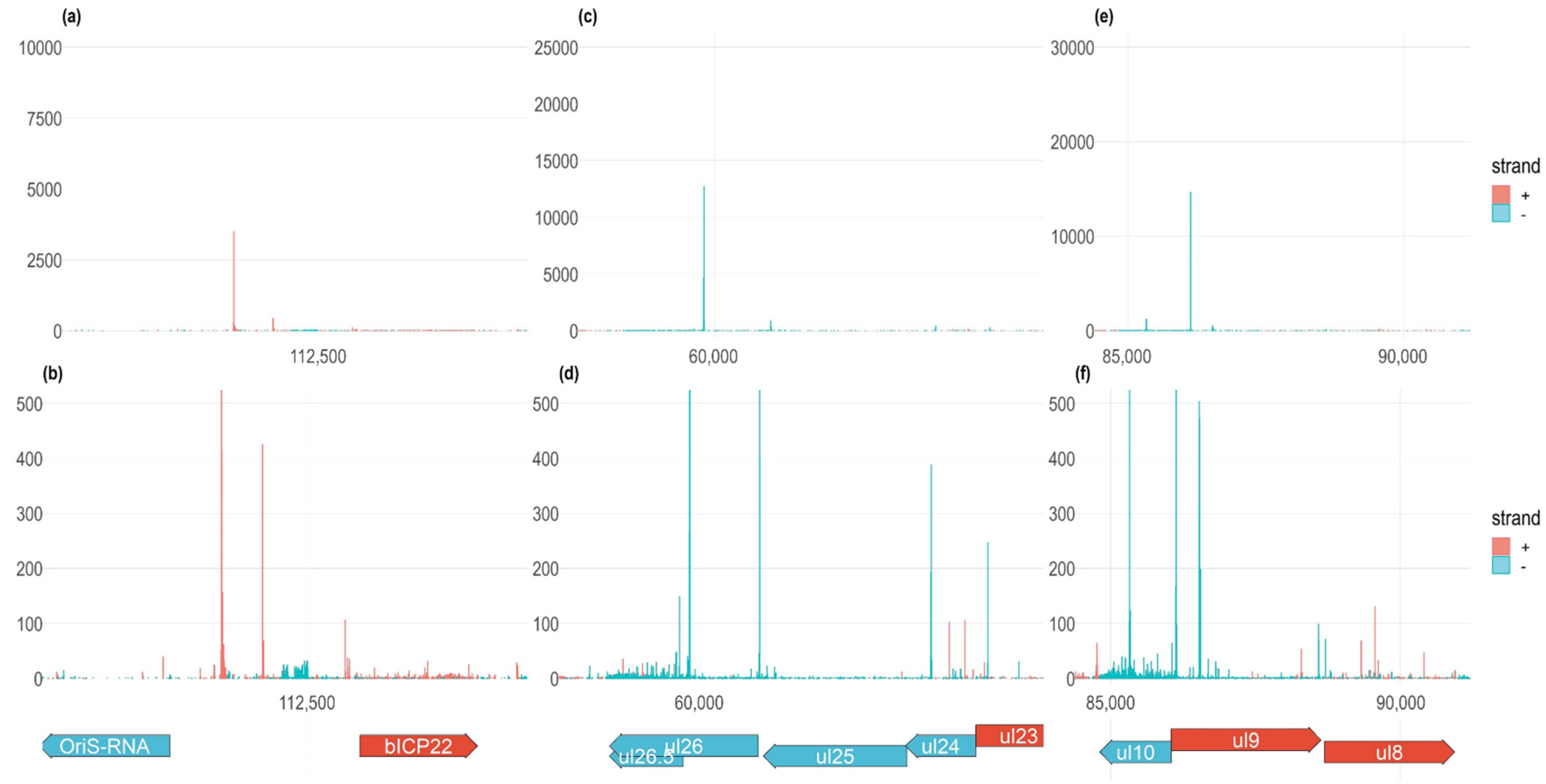
3.3. Transcription Start Sites
3.4. Transcription Start Site Clusters
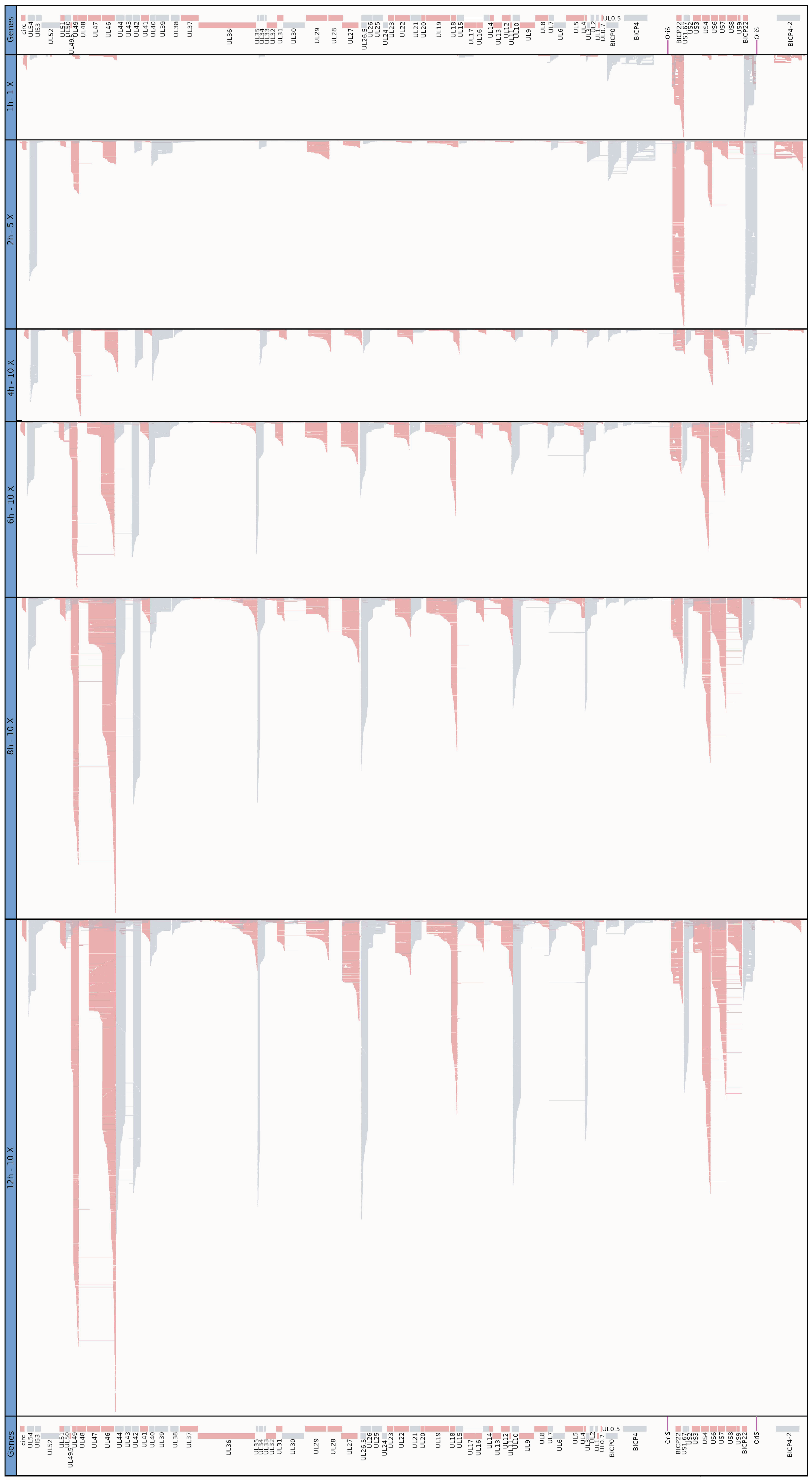
3.5. Time-Course Genome-Wide Expression of Transcription Start Sites
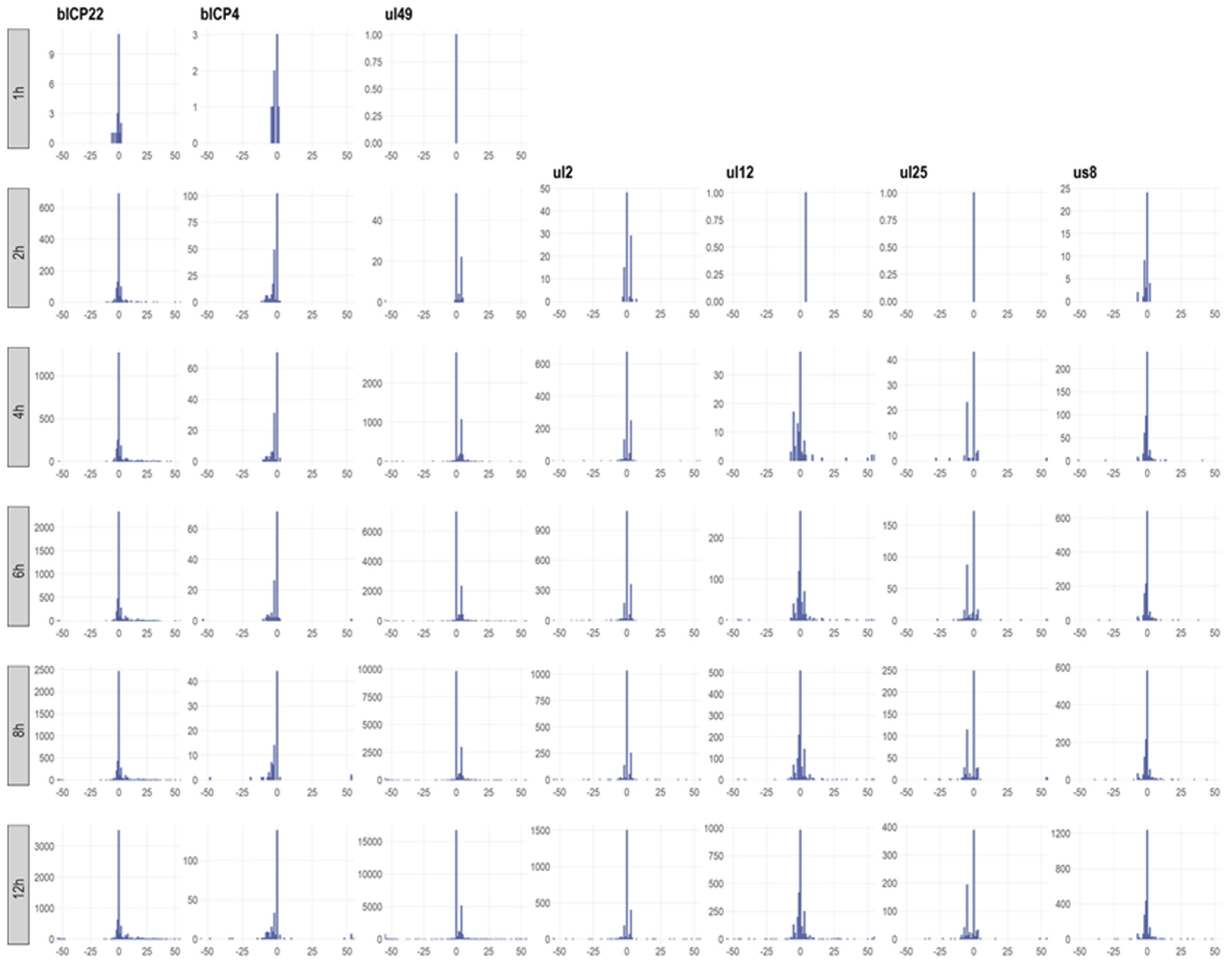
3.6. Transcription End Sites
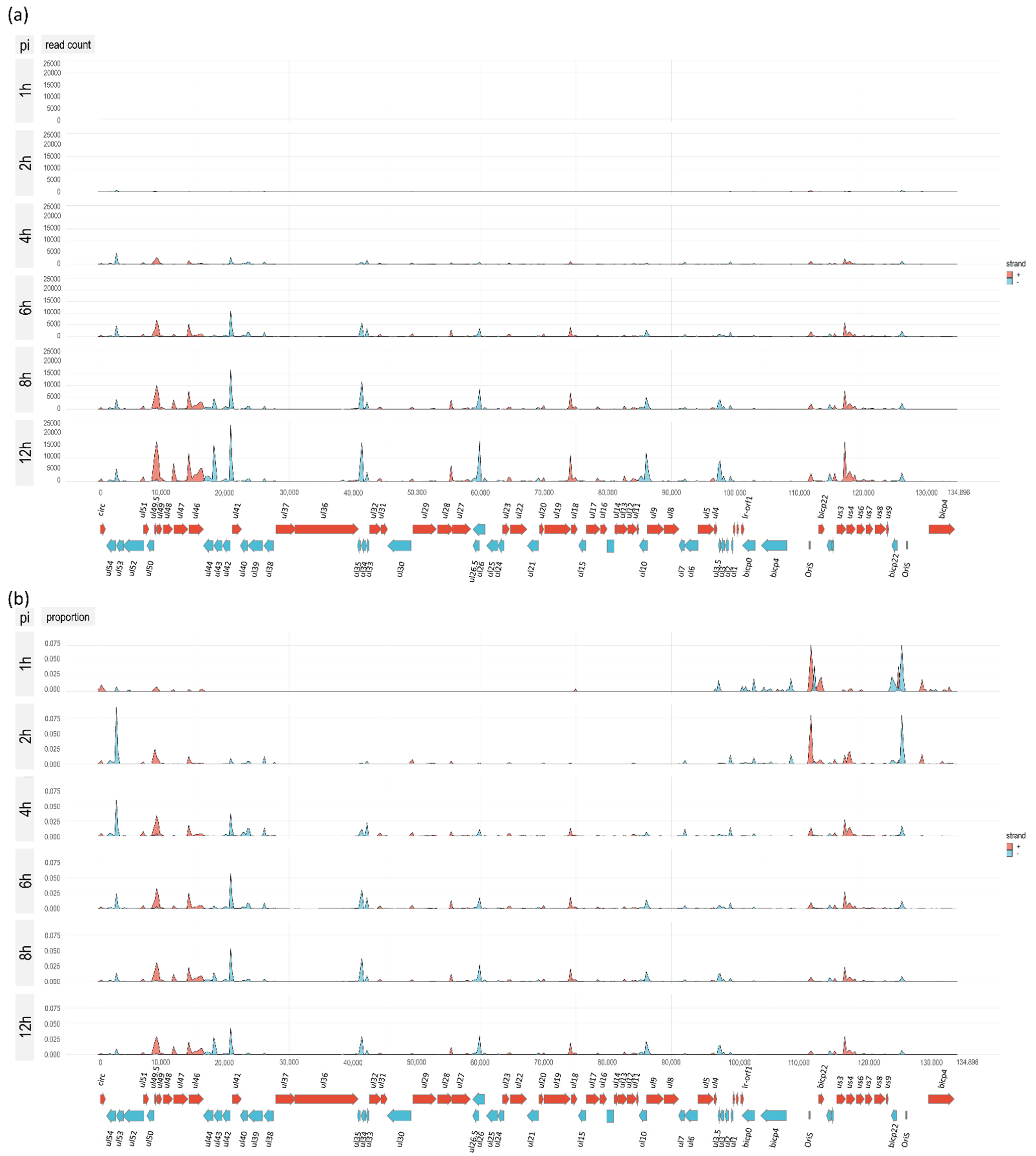
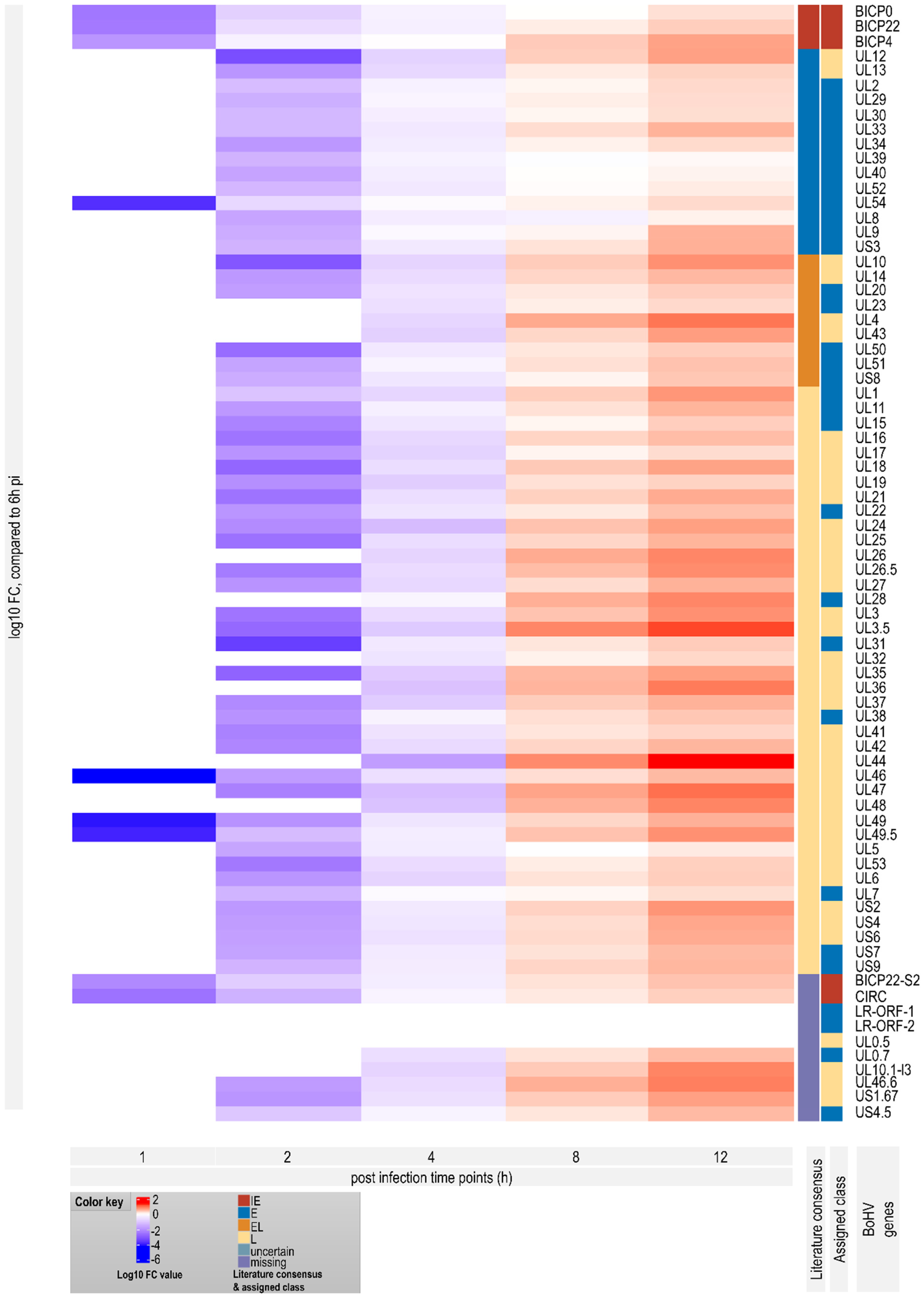
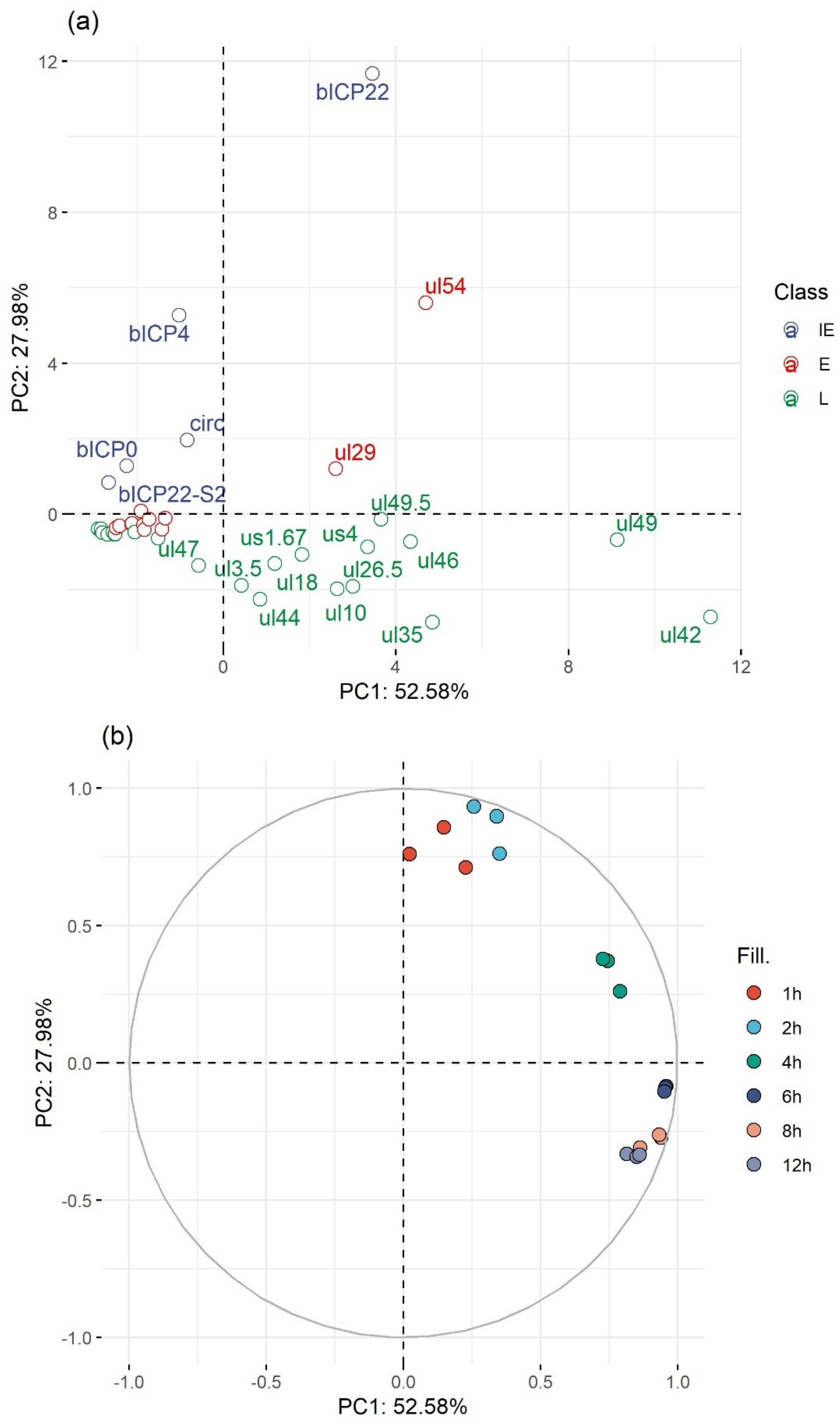
3.7. Genome-Wide Expression Dynamics of BoHV-1 Transcripts
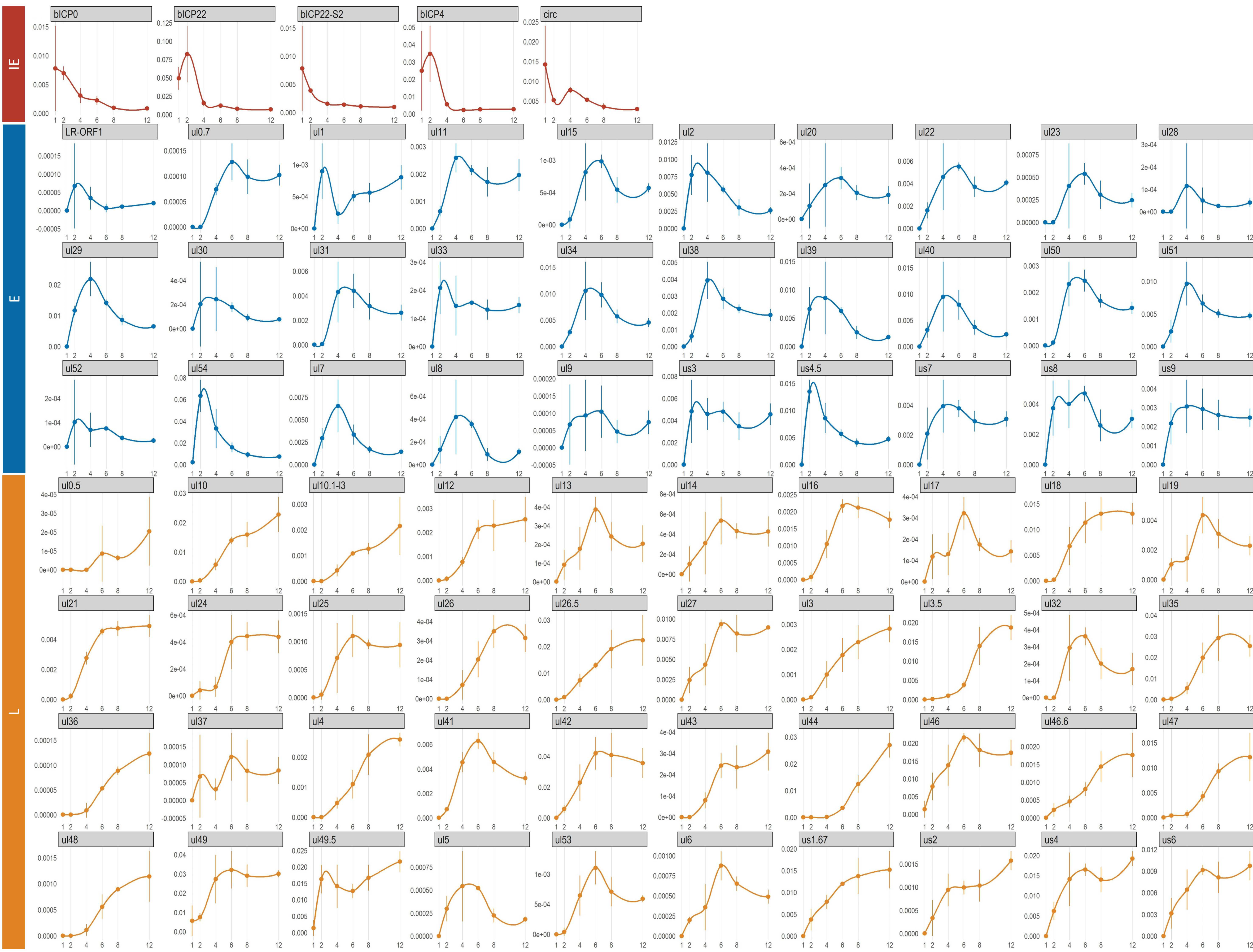
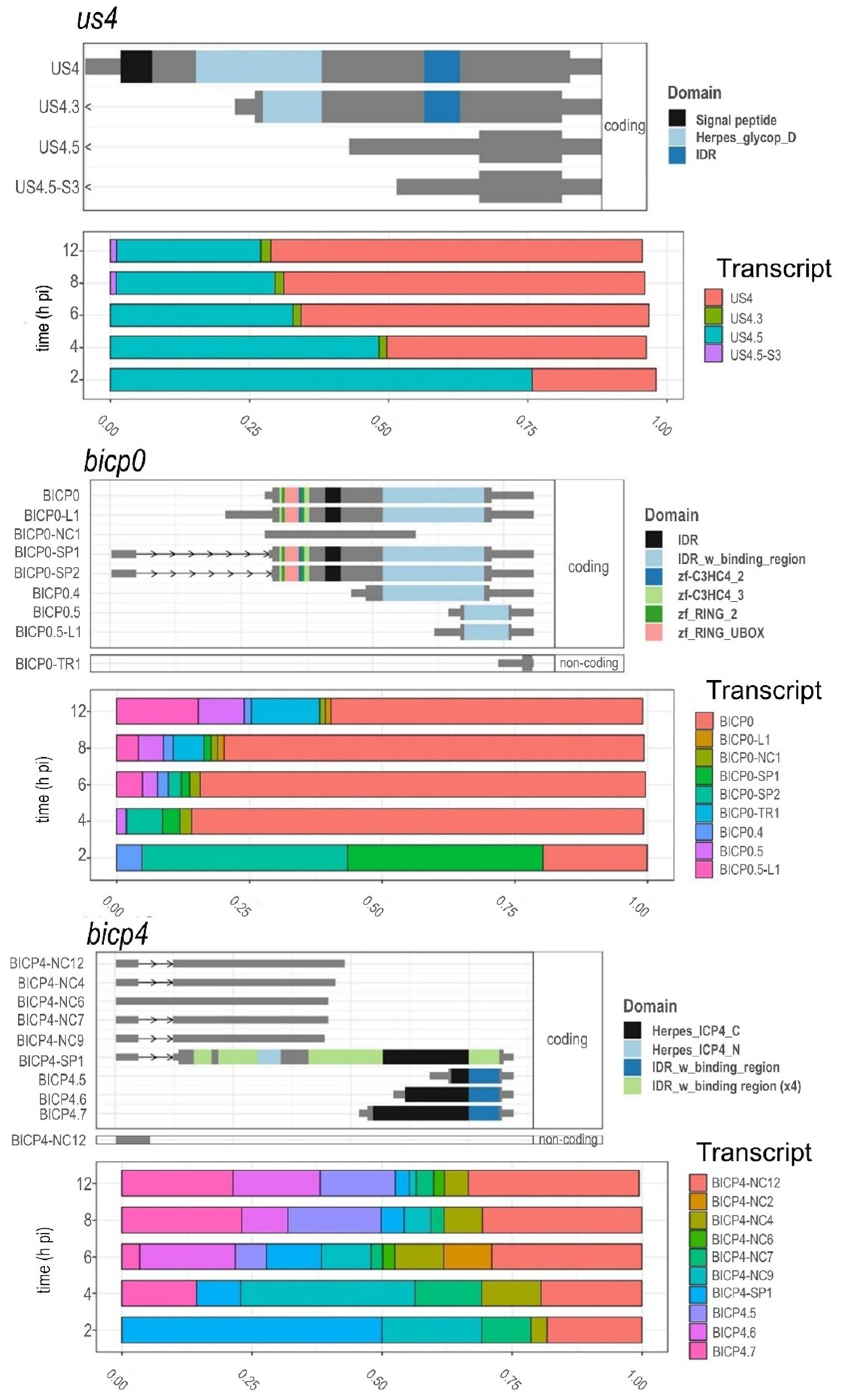
3.8. Time-Dependent Expression of Viral Gene Domains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Van Oirschot, J.T. Bovine herpesvirus 1 in semen of bulls and the risk of transmission: A brief review. Vet. Q. 1995, 17, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khattar, S.K.; Van Drunen Littel-Van Den Hurk, S.; Babiuk, L.A.; Tikoo, S.K. Identification and transcriptional analysis of a 3′-coterminal gene cluster containing UL1, UL2, UL3, and UL3.5 open reading frames of bovine herpesvirus-1. Virology 1995, 213, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlček, Č.; Benes, V.; Lu, Z.; Kutish, G.F.; Paces, V.; Rock, D.; Letchworth, G.J.; Schwyzer, M. Nucleotide sequence analysis of a 30-kb region of the bovine herpesvirus 1 genome which exhibits a colinear gene arrangement with the UL21 to UL4 genes of herpes simplex virus. Virology 1995, 210, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- d’Offay, J.M.; Fulton, R.W.; Eberle, R. Complete genome sequence of the NVSL BoHV-1.1 Cooper reference strain. Arch. Virol. 2013, 158, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- D’Offay, J.M.; Eberle, R.; Fulton, R.W.; Kirkland, P.D. Complete genomic sequence and comparative analysis of four genital and respiratory isolates of bovine herpesvirus subtype 1.2b (BoHV-1.2b), including the prototype virus strain K22. Arch. Virol. 2016, 161, 3269–3274. [Google Scholar] [CrossRef]
- Moldován, N.; Torma, G.; Gulyás, G.; Hornyák, Á.; Zádori, Z.; Jefferson, V.A.; Csabai, Z.; Boldogkői, M.; Tombácz, D.; Meyer, F.; et al. Time-course profiling of bovine herpesvirus type 1.1 transcriptome using multiplatform sequencing. Sci. Rep. 2020, 10, 20496. [Google Scholar] [CrossRef]
- Wirth, U.V.; Gunkel, K.; Engels, M.; Schwyzer, M. Spatial and temporal distribution of bovine herpesvirus 1 transcripts. J. Virol. 1989, 63, 4882–4889. [Google Scholar] [CrossRef] [Green Version]
- Wirth, U.V.; Fraefel, C.; Vogt, B.; Vlcek, C.; Paces, V.; Schwyzer, M. Immediate-early RNA 2.9 and early RNA 2.6 of bovine herpesvirus 1 are 3′- coterminal and encode a putative zinc finger transactivator protein. J. Virol. 1992, 66, 2763–2772. [Google Scholar] [CrossRef] [Green Version]
- Misra, V.; Walker, S.; Hayes, S.; O’Hare, P. The bovine herpesvirus α gene trans-inducing factor activates transcription by mechanisms different from those of its herpes simplex virus type 1 counterpart VP16. J. Virol. 1995, 69, 5209–5216. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Li, Q.; Jones, C. The bovine herpesvirus 1 regulatory proteins, bICP4 and bICP22, are expressed during the escape from latency. J. Neurovirol. 2019, 25, 42–49. [Google Scholar] [CrossRef]
- Köppel, R.; Vogt, B.; Schwyzer, M. Immediate-early protein BICP22 of bovine herpesvirus 1 trans-represses viral promoters of different kinetic classes and is itself regulated by BICP0 at transcriptional and posttranscriptional levels. Arch. Virol. 1997, 142, 2447–2464. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Wu, W.J.; Liu, L.D.; Wang, L.C.; Zhang, Y.; Wu, L.Q.; Guan, Y.; Li, Q.H. Herpes Simplex Virus 1 ICP22 Inhibits the Transcription of Viral Gene Promoters by Binding to and Blocking the Recruitment of P-TEFb. PLoS ONE 2012, 7, e45749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraefel, C.; Wirth, U.V.; Vogt, B.; Schwyzer, M. Immediate-early transcription over covalently joined genome ends of bovine herpesvirus 1: The circ gene. J. Virol. 1993, 67, 1328–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kronstad, L.M.; Brulois, K.F.; Jung, J.U.; Glaunsinger, B.A. Dual short upstream open reading frames control translation of a herpesviral polycistronic mRNA. PLoS Pathog. 2013, 9, e1003156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin. Microbiol. Rev. 2003, 16, 79–95. [Google Scholar] [CrossRef] [Green Version]
- Oláh, P.; Tombácz, D.; Csabai, Z.; Póka, N.; Prazsák, I.; Boldogkői, Z. Characterization of pseudorabies virus transcriptome by Illumina sequencing. BMC Microbiol. 2015, 15, 130. [Google Scholar] [CrossRef] [Green Version]
- Wade, J.T.; Grainger, D.C. Pervasive transcription: Illuminating the dark matter of bacterial transcriptomes. Nat. Rev. Microbiol. 2014, 12, 647–653. [Google Scholar] [CrossRef]
- Shiraki, T.; Kondo, S.; Katayama, S.; Waki, K.; Kasukawa, T.; Kawaji, H.; Kodzius, R.; Watahiki, A.; Nakamura, M.; Arakawa, T.; et al. Cap analysis gene expression for high-throughput analysis of transcriptional starting point and identification of promoter usage. Proc. Natl. Acad. Sci. USA 2003, 100, 15776–15781. [Google Scholar] [CrossRef] [Green Version]
- Batut, P.; Dobin, A.; Plessy, C.; Carninci, P.; Gingeras, T.R. High-fidelity promoter profiling reveals wide- spread alternative promoter usage and transposon-driven developmental gene expression. Genome Res. 2013, 23, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Kawaji, H.; Kasukawa, T.; Forrest, A.; Carninci, P.; Hayashizaki, Y. The FANTOM5 collection, a data series underpinning mammalian transcriptome atlases in diverse cell types. Sci. Data 2017, 4, 170113. [Google Scholar] [CrossRef]
- Gupta, I.; Collier, P.G.; Haase, B.; Mahfouz, A.; Joglekar, A.; Floyd, T.; Koopmans, F.; Barres, B.; Smit, A.B.; Sloan, S.A.; et al. Single-cell isoform RNA sequencing characterizes isoforms in thousands of cerebellar cells. Nat. Biotechnol. 2018, 36, 1197–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardwick, S.A.; Hu, W.; Joglekar, A.; Fan, L.; Collier, P.G.; Foord, C.; Balacco, J.; Lanjewar, S.; Sampson, M.M.; Koopmans, F.; et al. Single-nuclei isoform RNA sequencing unlocks barcoded exon connectivity in frozen brain tissue. Nat. Biotechnol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Glazov, E.A.; Horwood, P.F.; Assavalapsakul, W.; Kongsuwan, K.; Mitchell, R.W.; Mitter, N.; Mahony, T.J. Characterization of microRNAs encoded by the bovine herpesvirus 1 genome. J. Gen. Virol. 2010, 91, 32–41. [Google Scholar] [CrossRef]
- Byrne, A.; Beaudin, A.E.; Olsen, H.E.; Jain, M.; Cole, C.; Palmer, T. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat. Commun. 2017, 8, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.-Y.; Deng, F.; Jia, X.; Li, C.; Lai, S.-J. A transcriptome atlas of rabbit revealed by PacBio single-molecule long-read sequencing. Sci. Rep. 2017, 7, 7648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kakuk, B.; Kiss, A.A.; Torma, G.; Csabai, Z.; Prazsák, I.; Mizik, M.; Megyeri, K.; Tombácz, D.; Boldogkői, Z. Nanopore Assay Reveals Cell-Type-Dependent Gene Expression of Vesicular Stomatitis Indiana Virus and Differential Host Cell Response. Pathogens 2021, 10, 1196. [Google Scholar] [CrossRef]
- Moldován, N.; Balázs, Z.; Tombácz, D.; Csabai, Z.; Szűcs, A.; Snyder, M.; Boldogkői, Z. Multi-platform analysis reveals a complex transcriptome architecture of a circovirus. Virus Res. 2017, 237, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Nudelman, G.; Frasca, A.; Kent, B.; Sadler, K.C.; Sealfon, S.C.; Walsh, M.J.; Zaslavsky, E. High resolution annotation of zebrafish transcriptome using long-read sequencing. Genome Res. 2018, 28, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Prazsák, I.; Szucs, A.; Dénes, B.; Snyder, M.; Boldogkoi, Z. Dynamic transcriptome profiling dataset of vaccinia virus obtained from long-read sequencing techniques. Gigascience 2018, 7, giy139. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fang, C.; Fu, Y.; Hu, A.; Li, C.; Zou, C.; Li, X.; Zhao, S.; Zhang, C.; Li, C. A survey of transcriptome complexity in Sus scrofa using single-molecule long-read sequencing. DNA Res. 2018, 25, 421–437. [Google Scholar] [CrossRef]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Balázs, Z.; Kis, E.; Molnár, J.; Boldogkői, Z. Third-generation sequencing reveals extensive polycistronism and transcriptional overlapping in a baculovirus. Sci. Rep. 2018, 8, 8604. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zhang, H.; Kohnen, M.V.; Prasad, K.V.S.K.; Gu, L.; Reddy, A.S.N. Analysis of transcriptome and epitranscriptome in plants using PacBio iso-seq and nanopore-based direct RNA sequencing. Front. Genet. 2019, 10, 253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldogkői, Z.; Moldován, N.; Balázs, Z.; Snyder, M.; Tombácz, D. Long-read sequencing—A powerful tool in viral transcriptome research. Trends Microbiol. 2019, 27, 578–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, F.; Zhang, J.; Liu, Q.; Liu, X.; Wang, H.; He, J.; Kang, L. Long-read direct RNA sequencing by 5′-Cap capturing reveals the impact of Piwi on the widespread exonization of transposable elements in locusts. RNA Biol. 2019, 16, 950–959. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, J.; Wang, X.; Wei, Z. Full-length RNA sequencing reveals unique transcriptome composition in bermudagrass. Plant Physiol. Biochem. 2018, 132, 95–103. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Havelda, Z.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of novel transcripts in pseudorabies virus. Viruses 2015, 7, 2727–2744. [Google Scholar] [CrossRef] [Green Version]
- O’Grady, T.; Wang, X.; Höner Zu Bentrup, K.; Baddoo, M.; Concha, M.; Flemington, E.K. Global transcript structure resolution of high gene density genomes through multi-platform data integration. Nucleic Acids Res. 2016, 44, e145. [Google Scholar] [CrossRef]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Balázs, Z.; Likó, I.; Zsigmond, L.; Sharon, D.; Snyder, M.; Boldogkői, Z. Full-Length Isoform Sequencing Reveals Novel Transcripts and Substantial Transcriptional Overlaps in a Herpesvirus. PLoS ONE 2016, 11, e0162868. [Google Scholar] [CrossRef] [Green Version]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 2018, 8, 2708. [Google Scholar] [CrossRef] [Green Version]
- Balázs, Z.; Tombácz, D.; Csabai, Z.; Szűcs, A.; Megyeri, K.; Petrov, A.N.; Snyder, M.; Boldogkői, Z. Long-Read Sequencing of Human Cytomegalovirus Transcriptome Reveals RNA Isoforms Carrying Distinct Coding Potentials. Sci. Rep. 2017, 7, 15989. [Google Scholar] [CrossRef] [Green Version]
- Tombácz, D.; Balázs, Z.; Gulyás, G.; Csabai, Z.; Boldogkői, M.; Snyder, M.; Boldogkői, Z. Multiple Long-read Sequencing Survey of Herpes Simplex Virus Lytic Transcriptome. Front. Genet. 2019, 10, 834. [Google Scholar] [CrossRef] [PubMed]
- Depledge, D.P.; Srinivas, K.P.; Sadaoka, T.; Bready, D.; Mori, Y.; Placantonakis, D.G.; Mohr, I.; Wilson, A.C. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat. Commun. 2019, 10, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombácz, D.; Torma, G.; Gulyás, G.; Moldován, N.; Snyder, M.; Boldogkői, Z. Meta-analytic approach for transcriptome profiling of herpes simplex virus type 1. Sci. Data 2020, 7, 223. [Google Scholar] [CrossRef] [PubMed]
- Haberle, V.; Stark, A. Eukaryotic core promoters and the functional basis of transcription initiation. Nat. Rev. Mol. Cell. Biol. 2018, 19, 621–637. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Prazsák, I.; Csabai, Z.; Moldován, N.; Balázs, Z.; Dénes, B.; Snyder, M.; Boldogkői, Z. Long-read Assays Shed New Light on the Transcriptome Complexity of a Viral Pathogen. Sci. Rep. 2020, 10, 13822. [Google Scholar] [CrossRef]
- Kakuk, B.; Tombácz, D.; Balázs, Z.; Moldován, N.; Csabai, Z.; Torma, G.; Megyeri, K.; Snyder, M.; Boldogkői, Z. Combined Nanopore and Single-Molecule Real-Time Sequencing Survey of Human Betaherpesvirus 5 Transcriptome. Sci. Rep. 2021, 11, 14487. [Google Scholar] [CrossRef]
- Torma, G.; Tombácz, D.; Csabai, Z.; Moldován, N.; Mészáros, I.; Zádori, Z.; Boldogkői, Z. Combined Short and Long-read Sequencing Reveals a Complex Transcriptomic Architecture of African Swine Fever Virus. Viruses 2021, 13, 579. [Google Scholar] [CrossRef]
- Fülöp, Á.; Torma, G.; Moldován, N.; Szenthe, K.; Bánáti, F.; Almsarrhad, I.A.; Csabai, Z.; Tombácz, D.; Minárovits, J.; Boldogkői, Z. Integrative Profiling of Epstein-Barr Virus Transcriptome Using a Multiplatform Approach. Virol. J. 2022, 19, 7. [Google Scholar] [CrossRef]
- Hampsey, M. Molecular genetics of the RNA polymerase II general transcriptional machinery. Microbiol. Mol. Biol. Rev. 1998, 62, 465–503. [Google Scholar] [CrossRef] [Green Version]
- Carninci, P.; Sandelin, A.; Lenhard, B.; Katayama, S.; Shimokawa, K.; Ponjavic, J.; Semple, C.A.; Taylor, M.S.; Engström, P.G.; Frith, M.C.; et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat. Genet. 2006, 38, 626–635. [Google Scholar] [CrossRef]
- Ni, T.; Corcoran, D.L.; Rach, E.A.; Song, S.; Spana, E.P.; Gao, Y.; Ohler, U.; Zhu, J. A paired-end sequencing strategy to map the complex landscape of transcription initiation. Nat. Methods. 2010, 7, 521–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FANTOM Consortium and the RIKEN PMI and CLST (DGT); Forrest, A.R.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.L.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombácz, D.; Balázs, Z.; Csabai, Z.; Moldován, N.; Szűcs, A.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of the Dynamic Transcriptome of a Herpesvirus with Long-read Single Molecule Real-Time Sequencing. Sci. Rep. 2017, 7, 43751. [Google Scholar] [CrossRef] [PubMed]
- Balázs, Z.; Tombácz, D.; Csabai, Z.; Moldován, N.; Snyder, M.; Boldogkői, Z. Template-switching artifacts resemble alternative polyadenylation. BMC Genom. 2019, 20, 824. [Google Scholar] [CrossRef] [Green Version]
- Sessegolo, C.; Cruaud, C.; Da Silva, C.; Cologne, A.; Dubarry, M.; Derrien, T.; Lacroix, V.; Aury, J.M. Transcriptome profiling of mouse samples using nanopore sequencing of cDNA and RNA molecules. Sci. Rep. 2019, 9, 14908. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Tombácz, D.; Moldován, N.; Torma, G.; Nagy, T.; Hornyák, Á.; Csabai, Z.; Gulyás, G.; Boldogkői, M.; Jefferson, V.A.; Zádori, Z.; et al. Dynamic Transcriptome Sequencing of Bovine Alphaherpesvirus Type 1 and Host Cells Carried Out by a Multi-Technique Approach. Front. Genet. 2021, 7, 619056. [Google Scholar] [CrossRef]
- Morgan, M.; Pagès, H.; Obenchain, V.; Hayden, N. Rsamtools: Binary alignment (BAM), FASTA, variant call (BCF), and tabix file import. R Package Version 2.10.0 2021, 1, 677–689. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Reyes, A.; Huber, W. Detecting differential usage of exons from RNA-seq data. Genome Res. 2012, 22, 4025. [Google Scholar] [CrossRef]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Mészáros, B.; Erdős, G.; Dosztányi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Wirth, U.V.; Vogt, B.; Schwyzer, M. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J. Virol. 1991, 65, 195–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; et al. The Role of VP16 in the Life Cycle of Alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef] [PubMed]
- Some Genetic Functions Encoded by Herpes Simplex Virus Type 1. Available online: http://darwin.bio.uci.edu/~faculty/wagner/table.html (accessed on 3 October 2003).
- The Genetic and Transcription Map of the HSV-1 Genome. Available online: http://darwin.bio.uci.edu/~faculty/wagner/hsvimg04z.jpg (accessed on 3 October 2003).
- Roizman, B. The function of herpes simplex virus genes: A primer for genetic engineering of novel vectors. Proc. Nat. Acad. Sci. USA 1996, 93, 11307–11312. [Google Scholar] [CrossRef] [Green Version]
- Roizman, B.; Campadelli-Fiume, G. Alphaherpes viral genes and their functions. In Human Herpesviruses-Biology, Therapy and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; pp. 70–92. [Google Scholar]
- Pseudorabies Virus Gene Annotations. Available online: https://www.fli.de/en/institutes/institute-of-molecular-virology-and-cell-biology-imvz/laboratories/laboratory-for-virus-host-cell-interactions/fig-7-gene-and-transcript-arrangement-in-the-prv-genome/ (accessed on 1 September 2005).
- Vitting-Seerup, K.; Sandelin, A. The landscape of isoform switches in human cancers. Mol. Cancer Res. 2017, 15, 1206–1220. [Google Scholar] [CrossRef] [Green Version]
- Prazsák, I.; Moldován, N.; Balázs, Z.; Tombácz, D.; Megyeri, K.; Szűcs, A.; Csabai, Z.; Boldogkői, Z. Long-read sequencing uncovers a complex transcriptome topology in varicella zoster virus. BMC Genom. 2018, 19, 873. [Google Scholar] [CrossRef] [Green Version]
- Mathieu-Daudé, F.; Welsh, J.; Vogt, T.; McClelland, M. DNA rehybridization during PCR: The ‘Cot Effect’ and its consequences. Nucleic Acids Res. 1996, 24, 2080–2086. [Google Scholar] [CrossRef] [Green Version]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Taira, H.; Tsunoda, T.; Mizushima-Sugano, J.; Sese, J.; Hata, H.; Ota, T.; Isogai, T.; Tanaka, T.; Morishita, S.; et al. Diverse transcriptional initiation revealed by fine, large-scale mapping of mRNA start sites. EMBO Rep. 2001, 2, 388–393. [Google Scholar] [CrossRef] [Green Version]
- Smale, S.T.; Kadonaga, J.T. The RNA polymerase II core promoter. Annu. Rev. Biochem. 2003, 72, 449–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Park, J.K.; Zhang, J.Z. Evidence that alternative transcriptional initiation is largely nonadaptive. PLoS Biol. 2019, 17, e3000197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaji, H.; Frith, M.C.; Katayama, S.; Sandelin, A.; Kai, C.; Kawai, J.; Carninci, P.; Hayashizaki, Y. Dynamic usage of transcription start sites within core promoters. Genome Biol. 2006, 7, R118. [Google Scholar] [CrossRef] [Green Version]
- Barber, K.A.; Daugherty, H.C.; Ander, S.E.; Jefferson, V.A.; Shack, L.A.; Pechan, T.; Nanduri, B.; Meyer, F. Protein Composition of the Bovine Herpesvirus 1.1 Virion. Vet Sci. 2017, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokhriyal, M.; Ratta, B.; Yadav, B.S.; Kumar, A.; Saxena, M.; Verma, O.P.; Sharma, B. Three newly identified Immediate Early Genes of Bovine herpesvirus 1 lack the characteristic Octamer binding motif-1. Sci. Rep. 2018, 8, 11441. [Google Scholar] [CrossRef] [PubMed]
- Boldogkői, Z.; Balázs, Z.; Moldován, N.; Prazsák, I.; Tombácz, D. Novel classes of replication-associated transcripts discovered in viruses. RNA Biol. 2019, 16, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldogkői, Z.; Tombácz, D.; Balázs, Z. Interactions between the Transcription and Replication Machineries Regulate the RNA and DNA synthesis in the Herpesviruses. Virus Genes 2019, 55, 274–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boldogkői, Z. Transcriptional interference networks coordinate the expression of functionally-related genes clustered in the same genomic loci. Front. Genet. 2012, 3, 122. [Google Scholar] [CrossRef] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time Point | Barcode | Barcode Sequence | |
|---|---|---|---|
| 1st replicate | 1 h | A1(BC01) | AAGAAAGTTGTCGGTGTCTTTGTG |
| 2 h | A2(BC02) | TCGATTCCGTTTGTAGTCGTCTGT | |
| 4 h | A3(BC03) | GAGTCTTGTGTCCCAGTTACCAGG | |
| 6 h | A4(BC04) | TTCGGATTCTATCGTGTTTCCCTA | |
| 8 h | A5(BC05) | CTTGTCCAGGGTTTGTGTAACCTT | |
| 12 h | A6(BC06) | TTCTCGCAAAGGCAGAAAGTAGTC | |
| MOCK | A7(BC07) | GTGTTACCGTGGGAATGAATCCTT | |
| 2nd replicate | 1 h | A8(BC08) | TTCAGGGAACAAACCAAGTTACGT |
| 2 h | A9(BC09) | AACTAGGCACAGCGAGTCTTGGTT | |
| 4 h | A10(BC10) | AAGCGTTGAAACCTTTGTCCTCTC | |
| 6 h | A11(BC11) | GTTTCATCTATCGGAGGGAATGGA | |
| 8 h | A12(BC24) | GCATAGTTCTGCATGATGGGTTAG | |
| 12 h | A1(BC01) | AAGAAAGTTGTCGGTGTCTTTGTG | |
| MOCK | A2(BC02) | TCGATTCCGTTTGTAGTCGTCTGT | |
| 3rd replicate | 1 h | A3(BC03) | GAGTCTTGTGTCCCAGTTACCAGG |
| 2 h | A4(BC04) | TTCGGATTCTATCGTGTTTCCCTA | |
| 4 h | A5(BC05) | CTTGTCCAGGGTTTGTGTAACCTT | |
| 6 h | A6(BC06) | TTCTCGCAAAGGCAGAAAGTAGTC | |
| 8 h | A7(BC07) | GTGTTACCGTGGGAATGAATCCTT | |
| 12 h | A8(BC08) | TTCAGGGAACAAACCAAGTTACGT | |
| MOCK | A9(BC09) | AACTAGGCACAGCGAGTCTTGGTT |
| Gene | Sequence Names | Start | End | 6 h 20 mg | 8 h 20 mg | 6 h 100 mg | 8 h 100 mg |
|---|---|---|---|---|---|---|---|
| bicp22 | JX898220.1 | 112,888 | 113,790 | 132,263 | 165,542 | 138,780 | 127,408 |
| bicp4 | JX898220.1 | 103,907 | 107,941 | 44,292 | 57,067 | 36,249 | 43,288 |
| circ | JX898220.1 | 487 | 1227 | 5781 | 8667 | 6137 | 5398 |
| bicp0 | JX898220.1 | 100,898 | 102,949 | 4459 | 5270 | 4529 | 4234 |
| ul54 | JX898220.1 | 1648 | 2850 | 13,953 | 62,679 | 4601 | 4841 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tombácz, D.; Kakuk, B.; Torma, G.; Csabai, Z.; Gulyás, G.; Tamás, V.; Zádori, Z.; Jefferson, V.A.; Meyer, F.; Boldogkői, Z. In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing. Viruses 2022, 14, 1289. https://doi.org/10.3390/v14061289
Tombácz D, Kakuk B, Torma G, Csabai Z, Gulyás G, Tamás V, Zádori Z, Jefferson VA, Meyer F, Boldogkői Z. In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing. Viruses. 2022; 14(6):1289. https://doi.org/10.3390/v14061289
Chicago/Turabian StyleTombácz, Dóra, Balázs Kakuk, Gábor Torma, Zsolt Csabai, Gábor Gulyás, Vivien Tamás, Zoltán Zádori, Victoria A. Jefferson, Florencia Meyer, and Zsolt Boldogkői. 2022. "In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing" Viruses 14, no. 6: 1289. https://doi.org/10.3390/v14061289