
The Genetic Characterization of a Novel Natural Recombinant Pseudorabies Virus in China

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation of PRV FJ Strain
2.2. Concentration and Purification of the Virion
2.3. DNA Extraction
- (1)
- 200 μL of the 10% SDS solution, and 15 μL of the 10 mg/mL RNase A were added in the Eppendorf tubes and incubated at 60 °C for 30 min in the metal bath (Cole-Parmer, Chicago, IL, USA).
- (2)
- 100 μL of the 10 mg/mL Proteinase K was added and incubated in a metal bath at 56 °C for 30 min.
- (3)
- The ddH2O was added to make up the concentrated viral solution to 400 μL, then 600 μL of phenol: chloroform: isoamyl alcohol = 25:24:1 DNA extraction reagent was added, the Eppendorf tubes were carefully inverted and mixed, then the Eppendorf tubes were stood for 5 min to make the liquid stratified.
- (4)
- These Eppendorf tubes were centrifuged for 5 min at 12,000 r/min with a microcentrifuge (Thermo Scientific, Waltham, MA, USA), and they took the supernatant to avoid aspirating to the impurities in the middle layer.
- (5)
- Repeat steps (3) and (4).
- (6)
- An equal volume of isopropanol was added, mixed lightly, and precipitated for 1 h in a refrigerator at −20 °C.
- (7)
- These Eppendorf tubes were centrifuged at 12,000 r/min for 5 min with the microcentrifuge to discard the supernatant, 800 μL of anhydrous ice ethanol was added, then 1/10 volume of 3 mol/L NaAc was added, washed with light mixing, and left for 5 min.
- (8)
- These Eppendorf tubes were centrifuged at 12,000 r/min for 5 min with the microcentrifuge at 4 °C to discard the supernatant, the precipitate was placed in a biosafety cabinet, the exhaust air was turned on and blown until there was no smell of alcohol.
- (9)
- The precipitate was carefully dissolved in 100μL TE solution.
2.4. Sequencing the Complete Genomes
2.5. Genome and Related Gene Homology and Phylogenetic Analysis
2.6. Prediction of Potential Genome Recombination Events
2.7. Sequence Submission
3. Results
3.1. Complete Genome Sequence Analysis
3.2. Genomic Genetic Evolution Analysis
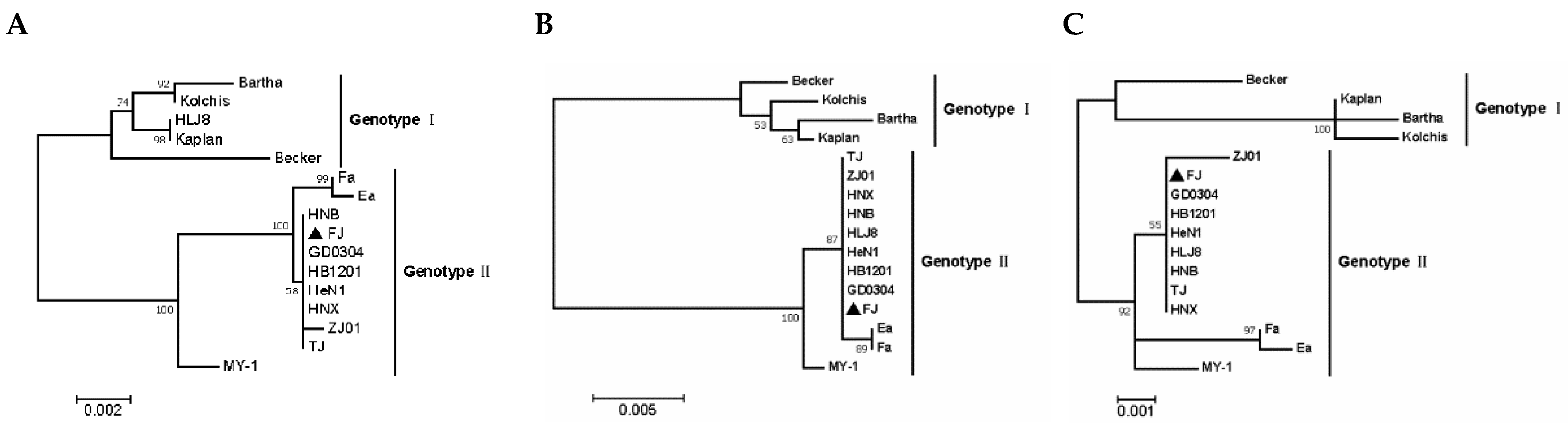
3.3. Phylogenetic Analysis of Related Gene Sequences
3.4. Recombination Analyses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aujeszky, A. Über Eine Neue Infektionskrankheit bei Haustieren; Fischer: Berlin, Germany, 1902; Volume 32. [Google Scholar]
- Lee, J.; Wilson, M. A review of pseudorabies (Aujeszky’s disease) in pigs. Can. Vet. J. 1979, 20, 65. [Google Scholar] [PubMed]
- Szpara, M.L.; Tafuri, Y.R.; Parsons, L.; Shamim, S.R.; Verstrepen, K.J.; Legendre, M.; Enquist, L. A wide extent of inter-strain diversity in virulent and vaccine strains of alphaherpesviruses. PLoS Pathog. 2011, 7, e1002282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Hahn, E.; Tottewitz, F.; Kramer, M.; Klupp, B.; Mettenleiter, T.; Freuling, C. Pseudorabies virus in wild swine: A global perspective. Arch. Virol. 2011, 156, 1691. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Hengartner, C.J.; Mettenleiter, T.C.; Enquist, L.W. Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 2004, 78, 424–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2011; Volume 9. [Google Scholar]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. MMBR 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauwynck, H.; Glorieux, S.; Favoreel, H.; Pensaert, M. Cell biological and molecular characteristics of pseudorabies virus infections in cell cultures and in pigs with emphasis on the respiratory tract. Vet. Res. 2007, 38, 229–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, T.-Q.; Peng, J.-M.; Tian, Z.-J.; Zhao, H.-Y.; Li, N.; Liu, Y.-M.; Chen, J.-Z.; Leng, C.-L.; Sun, Y.; Chang, D. Pseudorabies virus variant in Bartha-K61–vaccinated pigs, China, 2012. Emerg. Infect. Dis. 2013, 19, 1749. [Google Scholar] [CrossRef]
- Sun, Y.; Luo, Y.; Wang, C.-H.; Yuan, J.; Li, N.; Song, K.; Qiu, H.-J. Control of swine pseudorabies in China: Opportunities and limitations. Vet. Microbiol. 2016, 183, 119–124. [Google Scholar] [CrossRef]
- Yu, X.; Zhou, Z.; Hu, D.; Zhang, Q.; Han, T.; Li, X.; Gu, X.; Yuan, L.; Zhang, S.; Wang, B. Pathogenic pseudorabies virus, China, 2012. Emerg. Infect. Dis. 2014, 20, 102. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-Y.; Sun, Z.; Tan, F.-F.; Guo, L.-H.; Wang, Y.-Z.; Wang, J.; Wang, Z.-Y.; Wang, L.-L.; Li, X.-D.; Xiao, Y. Pathogenicity of a currently circulating Chinese variant pseudorabies virus in pigs. World J. Virol. 2016, 5, 23. [Google Scholar] [CrossRef]
- Ye, C.; Zhang, Q.-Z.; Tian, Z.-J.; Zheng, H.; Zhao, K.; Liu, F.; Guo, J.-C.; Tong, W.; Jiang, C.-G.; Wang, S.-J. Genomic characterization of emergent pseudorabies virus in China reveals marked sequence divergence: Evidence for the existence of two major genotypes. Virology 2015, 483, 32–43. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Li, N.; Cong, X.; Wang, C.-H.; Du, M.; Li, L.; Zhao, B.; Yuan, J.; Liu, D.-D.; Li, S. Pathogenicity and genomic characterization of a pseudorabies virus variant isolated from Bartha-K61-vaccinated swine population in China. Vet. Microbiol. 2014, 174, 107–115. [Google Scholar] [CrossRef]
- Yu, T.; Chen, F.; Ku, X.; Fan, J.; Zhu, Y.; Ma, H.; Li, S.; Wu, B.; He, Q. Growth characteristics and complete genomic sequence analysis of a novel pseudorabies virus in China. Virus Genes 2016, 52, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chen, F.; Ku, X.; Zhu, Y.; Ma, H.; Li, S.; He, Q. Complete genome sequence of novel pseudorabies virus strain HNB isolated in China. Genome Announc. 2016, 4, e01641-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, C.; Zhao, K.; Guo, J.; Jiang, C.; Chang, X.; Wang, S.; Wang, T.; Peng, J.; Cai, X.; Tian, Z. Genomic sequencing and genetic diversity analysis of a pseudorabies virus based on main infection or virulence genes. Zhongguo Yufang Shouyi Xuebao/Chin. J. Prev. Vet. Med. 2015, 37, 581–584. [Google Scholar]
- Mettenleiter, T.C.; Lukàcs, N.; Thiel, H.-J.; Schreurs, C.; Rziha, H.-J. Location of the structural gene of pseudorabies virus glycoprotein complex gII. Virology 1986, 152, 66–75. [Google Scholar] [CrossRef]
- Eloit, M.; Vannier, P.; Hutet, E.; Fournier, A. Correlation between gI, gII, gIII, and gp 50 antibodies and virus excretion in vaccinated pigs infected with pseudorabies virus. Arch. Virol. 1992, 123, 135–143. [Google Scholar] [CrossRef]
- Jöns, A.; Dijkstra, J.M.; Mettenleiter, T.C. Glycoproteins M and N of pseudorabies virus form a disulfide-linked complex. J. Virol. 1998, 72, 550–557. [Google Scholar] [CrossRef] [Green Version]
- Kit, S.; Kit, M.; Pirtle, E. Attenuated properties of thymidine kinase-negative deletion mutant of pseudorabies virus. Am. J. Vet. Res. 1985, 46, 1359–1367. [Google Scholar]
- Lipowski, A. Evaluation of efficacy and safety of Aujeszky’s. Pol. J. Vet. Sci. 2006, 9, 75–79. [Google Scholar] [PubMed]
- Schwartz, J.A.; Brittle, E.E.; Reynolds, A.E.; Enquist, L.W.; Silverstein, S.J. UL54-null pseudorabies virus is attenuated in mice but productively infects cells in culture. J. Virol. 2006, 80, 769–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, R.-Y.; Hsu, T.-W.; Chen, Y.-L.; Liu, S.-F.; Tsai, Y.-J.; Lin, Y.-T.; Chen, Y.-S.; Fan, Y.-H. Japanese encephalitis virus non-coding RNA inhibits activation of interferon by blocking nuclear translocation of interferon regulatory factor 3. Vet. Microbiol. 2013, 166, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Alonso, J.A.; Patterson, J.L. Sequence variability in viral genome non-coding regions likely contribute to observed differences in viral replication amongst MARV strains. Virology 2013, 440, 51–63. [Google Scholar] [CrossRef] [Green Version]
- Landeras-Bueno, S.; Ortin, J. Regulation of influenza virus infection by long non-coding RNAs. Virus Res. 2016, 212, 78–84. [Google Scholar] [CrossRef]
- Iwakiri, D. Multifunctional non-coding Epstein–Barr virus encoded RNAs (EBERs) contribute to viral pathogenesis. Virus Res. 2016, 212, 30–38. [Google Scholar] [CrossRef]
- Glazenburg, K.; Moormann, R.; Kimman, T.; Gielkens, A.; Peeters, B. Genetic recombination of pseudorabies virus: Evidence that homologous recombination between insert sequences is less frequent than between autologous sequences. Arch. Virol. 1995, 140, 671–685. [Google Scholar] [CrossRef]
- Da Silva, L.F.; Jones, C. Small non-coding RNAs encoded within the herpes simplex virus type 1 latency associated transcript (LAT) cooperate with the retinoic acid inducible gene I (RIG-I) to induce beta-interferon promoter activity and promote cell survival. Virus Res. 2013, 175, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Strain Name | Accession Number | Country | Isolation Date |
|---|---|---|---|---|
| 1 | Becker | JF797219.1 | USA | 1970 |
| 2 | Bartha | JF797217.1 | Hungary | 1961 |
| 3 | Ea | KU315430.1 | China | 1990 |
| 4 | Fa | KM189913.1 | China | 2012 |
| 5 | GD0304 | MH582511.1 | China | 2015 |
| 6 | HB1201 | KU057086.1 | China | 2012 |
| 7 | HeN1 | KP098534.1 | China | 2012 |
| 8 | HLJ8 | KT824771.1 | China | 2012 |
| 9 | HNB | KM189914.3 | China | 2012 |
| 10 | HNX | KM189912.1 | China | 2012 |
| 11 | Kaplan | JF797218.1 | Hungary | 1959 |
| 12 | Kolchis | KT983811.1 | Greece | 2010 |
| 13 | MY-1 | AP018925.1 | Japan | 2015 |
| 14 | ZJ-01 | KM061380.1 | China | 2012 |
| 15 | TJ | KJ789182.1 | China | 2012 |
| Region | Location | Length (bp) |
|---|---|---|
| UL | 1–101,012 | 101,012 |
| IRs | 101,013–117,681 | 16,669 |
| US | 117,682–127,034 | 9353 |
| TRs | 127,035–143,703 | 16,669 |
| Protein Name | Location of ORF (bp) | Length (aa) | Function |
|---|---|---|---|
| UL56 | 754–1377 | 207 | Possibly vesicular trafficking |
| ICP27 | 1932–3017 | 361 | Gene regulation; early protein |
| gK | 3096–4034 | 312 | Viral glycoprotein K; type III membrane protein |
| UL52 | 3989–6895 | 968 | DNA replication; primase subunit of ULS/UL8/UL52 complex |
| UL51 | 6882–7613 | 243 | Tegument protein |
| dUTPase | 7812–8621 | 269 | dUTPase |
| gN | 8542–8841 | 99 | Glycoprotein N; type I membrane protein; complexed with gM |
| VP22 | 8879–9619 | 246 | Interacts with C-terminal domains of gE and gM; tegument protein |
| VP16 | 9683–10,924 | 413 | Gene regulation (transactivator); egress (secondary envelopment); tegument protein |
| VP13/14 | 11,034–13,250 | 738 | Viral egress (secondary envelopment); tegument protein |
| VP11/12 | 13,269–15,356 | 695 | Possibly gene regulation; tegument protein |
| gB | 15,905–18,649 | 914 | Viral entry (fusion); cell–cell spread; glycoprotein B; type I membrane protein |
| ICP18.5 | 18,520–20,688 | 722 | DNA cleavage and encapsulations (terminase); associated with UL15, UL33 and UL6 |
| ICP8 | 20,836–24,378 | 1180 | DNA replication-recombination; binds single-stranded DNA |
| UL30 | 24,677–27,823 | 1048 | DNA replication; DNA polymerase subunit of UL30/UL42 complex |
| UL31 | 27,744–28,559 | 271 | Viral egress (nuclear egress); primary virion tegument protein; interacts with UL34 |
| UL32 | 28,552–29,967 | 471 | DNA packaging; efficient localization of capsids to replication compartments |
| UL33 | 29,966–30,322 | 118 | DNA cleavage and encapsidation; associated with UL28 and UL 15 |
| UL34 | 30,494–31,279 | 261 | Viral egress (nuclear egress); primary virion envelop protein tail-anchored type II nuclear membrane protein; interacts with UL31 |
| VP26 | 31,334–31,645 | 103 | Capsid protein |
| VP1/2 | 32,057–41,644 | 3195 | Large tegument protein; interacts with UL37 and UL19 |
| UL37 | 41,682–44,441 | 919 | Tegument protein; interacts with UL36 |
| VP19c | 44,498–45,604 | 368 | Capsid protein; forms triplexes together with ULl8 |
| RR1 | 45,941–48,307 | 788 | Nucleotide synthesis; large subunit of ribonucleotide reductase |
| RR2 | 48,317–49,228 | 303 | Nucleotide synthesis; small subunit of ribonucleotide reductase |
| vhs | 49,843–50,940 | 365 | Gene regulation (inhibitor of gene expression); virion host cell shutoff |
| UL42 | 51,069–52,226 | 385 | DNA replication; polymerase accessory subunit of UL30/UL42 complex |
| UL43 | 52,286–53,407 | 373 | Unknown; type III membrane protein |
| gC | 53,474–54,937 | 487 | Viral entry (virion attachment); glycoprotein C; type I membrane protein; binds to heparan sulfate |
| UL26.5 | 55,233–56,093 | 286 | Scaffold protein; substrate for UL26; required for capsid formation and maturation |
| VP24 | 55,233–56,831 | 532 | Scaffold protein; proteinase; required for capsid formation and maturation |
| UL25 | 56,883–58,493 | 536 | Capsid-associated protein; required for capsid assembly |
| UL24 | 58,592–59,107 | 171 | Unknown; type III membrane protein |
| TK | 59,100–60,062 | 320 | Nucleotide synthesis; thymidine kinase |
| gH | 60,198–62,255 | 685 | Viral entry (fusion); cell–cell spread; glycoprotein H; type I membrane protein; complexed with gL |
| UL21 | 64,012–65,613 | 533 | Capsid-associated protein |
| UL20 | 65,720–66,217 | 165 | Viral egress; type III membrane protein |
| VP5 | 66,306–70,298 | 1330 | Major capsid protein; forms hexons and pentons |
| VP23 | 70,473–71,363 | 296 | Capsid protein; forms triplexes together with UL38 |
| UL17 | 72,739–74,538 | 599 | DNA cleavage and encapsidation |
| UL16 | 74,565–75,551 | 328 | Possibly virion morphogenesis |
| UL15 | 71,546–72,687 75,572–76,691 | 753 | DNA cleavage and encapsidation; terminase subunit; interacts with UL33 UL28, and UL6 |
| UL14 | 76,690–77,169 | 159 | Virion morphogenesis |
| VP18.8 | 77,139–78,314 | 391 | Protein-serine/threonine kinase |
| AN | 78,280–79,731 | 483 | DNA recombination; alkaline exonuclease |
| UL11 | 79,689–79,880 | 63 | Viral egress (secondary envelopment); membrane-associated tegument protein |
| gM | 80,309–81,490 | 393 | Viral egress (secondary envelopment); glycoprotein M; type III membrane protein; C terminus interacts with UL49; inhibits membrane fusion in transient assays; complexed with gN |
| OBP | 81,489–84,023 | 844 | Sequence-specific ori-binding protein |
| UL8 | 84,020–86,086 | 688 | DNA replication; part of ULS/UL8/UL52 helicase-primase complex |
| UL7 | 86,288–87,088 | 266 | Virion morphogenesis |
| UL6 | 86,979–88,916 | 645 | DNA packaging Capsid protein; portal protein; docking site for terminase |
| UL5 | 88,915–91,470 | 851 | DNA replication; part of ULS/LJL8/UL52 helicase-primase complex; helicase motif |
| UL4 | 91,528–91,965 | 145 | Nuclear protein |
| UL3.5 | 92,141–92,809 | 222 | Possibly virion morphogenesis |
| UL3 | 92,806–93,540 | 244 | Nuclear protein |
| UNG | 93,597–94,568 | 323 | Uracil-DNA glycosylase |
| gL | 94,546–95,016 | 156 | Viral entry; cell–cell spread; glycoprotein L; membrane-anchored via complex with gH |
| ICP0 | 96,248–97,348 | 366 | Gene regulation (transactivator of viral and cellular genes); early protein |
| ICP4 | 103,130–107,544 | 1471 | Gene regulation; immediate early protein |
| ICP22 | 116,146–117,339 | 397 | Gene regulation |
| PK | 118,467–119,471 | 334 | Minor form of protein kinase (53-kDa mobility); viral egress (nuclear egress); major form of protein kinase (41-kDa mobility) |
| gG | 119,531–121,030 | 499 | Cell–cell spread; secreted; glycoprotein G |
| gD | 121,214–122,422 | 402 | Viral entry (cellular receptor binding protein); glycoprotein D |
| gI | 122,446–123,543 | 465 | Cell–cell spread; glycoprotein I; type I membrane protein; complexed with gE |
| gE | 123,647–125,386 | 579 | Cell–cell spread; glycoprotein E; type I membrane protein; complexed with gI; C terminus interacts with UL49 |
| US9(11K) | 125,444–125,740 | 98 | Protein sorting in axons; type II tail-anchored membrane protein |
| US2(28K) | 125,994–126,764 | 256 | Possibly envelope associated |
| Virus Strain | Nucleotides Homology (%) | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MY-1 | FJ * | Bartha | Kaplan | Becker | TJ | ZJ01 | HNX | Fa | HNB | HeN1 | HLJ8 | Kolchis | HB1201 | Ea | GD0304 | |
| MY-1 | ||||||||||||||||
| FJ * | 99.0 | |||||||||||||||
| Bartha | 95.4 | 95.7 | ||||||||||||||
| Kaplan | 95.6 | 96.0 | 99.4 | |||||||||||||
| Becker | 95.4 | 95.7 | 98.6 | 98.9 | ||||||||||||
| TJ | 99.0 | 100 | 95.7 | 96.0 | 95.7 | |||||||||||
| ZJ01 | 99.0 | 99.9 | 95.6 | 95.9 | 95.6 | 99.9 | ||||||||||
| HNX | 99.0 | 100 | 95.7 | 96.0 | 95.7 | 100 | 99.9 | |||||||||
| Fa | 98.9 | 99.7 | 95.7 | 96.0 | 95.6 | 99.7 | 99.7 | 99.7 | ||||||||
| HNB | 99.0 | 100.0 | 95.7 | 96.0 | 95.7 | 100.0 | 99.9 | 100.0 | 99.7 | |||||||
| HeN1 | 99.0 | 100.0 | 95.7 | 96.0 | 95.7 | 100.0 | 99.9 | 100.0 | 99.7 | 100.0 | ||||||
| HLJ8 | 99.0 | 100.0 | 95.7 | 96.0 | 95.7 | 100.0 | 99.9 | 100.0 | 99.7 | 100.0 | 100.0 | |||||
| Kolchis | 95.4 | 95.7 | 99.1 | 99.5 | 99.0 | 95.7 | 95.6 | 95.7 | 95.7 | 95.7 | 95.7 | 95.7 | ||||
| HB1201 | 99.0 | 100.0 | 95.7 | 96.0 | 95.7 | 100.0 | 99.9 | 100.0 | 99.7 | 100.0 | 100.0 | 100.0 | 95.7 | |||
| Ea | 98.9 | 99.7 | 95.7 | 96.0 | 95.6 | 99.7 | 99.7 | 99.7 | 100.0 | 99.7 | 99.7 | 99.7 | 95.7 | 99.7 | ||
| GD0304 | 99.0 | 100.0 | 95.7 | 96.0 | 95.7 | 100.0 | 99.9 | 100.0 | 99.7 | 100.0 | 100.0 | 100.0 | 95.7 | 100.0 | 99.7 | |
| Position (bp) | Method and p Value | ||||||
|---|---|---|---|---|---|---|---|
| Bootscan | Maxchi | Chimaera | SiScan | PhylPro | LARD | 3Seq | |
| 1694–1936 | 2.31 × 10−11 * | 3.50 × 10−9 * | NS | 2.27 × 10−2 | 1.05 × 10−2 | NS | NS |
| 101,113–102,660 | 6.54 × 10−5 * | 6.26 × 10−3 | NS | 5.67 × 10−3 | 1.82 × 10−3 | NS | NS |
| 107,964–111,481 | 9.87 × 10−10 * | 9.81 × 10−9 * | NS | 1.93 × 10−5 * | 1.68 × 10−4 * | NS | NS |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Tang, W.; Wang, X.; Zhao, J.; Peng, K.; Sun, X.; Li, S.; Kuang, S.; Zhu, L.; Zhou, Y.; et al. The Genetic Characterization of a Novel Natural Recombinant Pseudorabies Virus in China. Viruses 2022, 14, 978. https://doi.org/10.3390/v14050978
Huang J, Tang W, Wang X, Zhao J, Peng K, Sun X, Li S, Kuang S, Zhu L, Zhou Y, et al. The Genetic Characterization of a Novel Natural Recombinant Pseudorabies Virus in China. Viruses. 2022; 14(5):978. https://doi.org/10.3390/v14050978
Chicago/Turabian StyleHuang, Jianbo, Wenjie Tang, Xvetao Wang, Jun Zhao, Kenan Peng, Xiangang Sun, Shuwei Li, Shengyao Kuang, Ling Zhu, Yuancheng Zhou, and et al. 2022. "The Genetic Characterization of a Novel Natural Recombinant Pseudorabies Virus in China" Viruses 14, no. 5: 978. https://doi.org/10.3390/v14050978