
Infectious Bronchitis Virus Nsp14 Degrades JAK1 to Inhibit the JAK-STAT Signaling Pathway in HD11 Cells

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Viruses, and Antibodies
2.2. Virus Infection Assay
2.3. Transfection and Dual-Luciferase Reporter Assays
2.4. Confocal Fluorescence Microscopy
2.5. Co-Immunoprecipitation Assays
2.6. Western Blot
2.7. Quantitative Real-Time Polymerase Chain Reaction
2.8. Statistical Analysis
3. Results
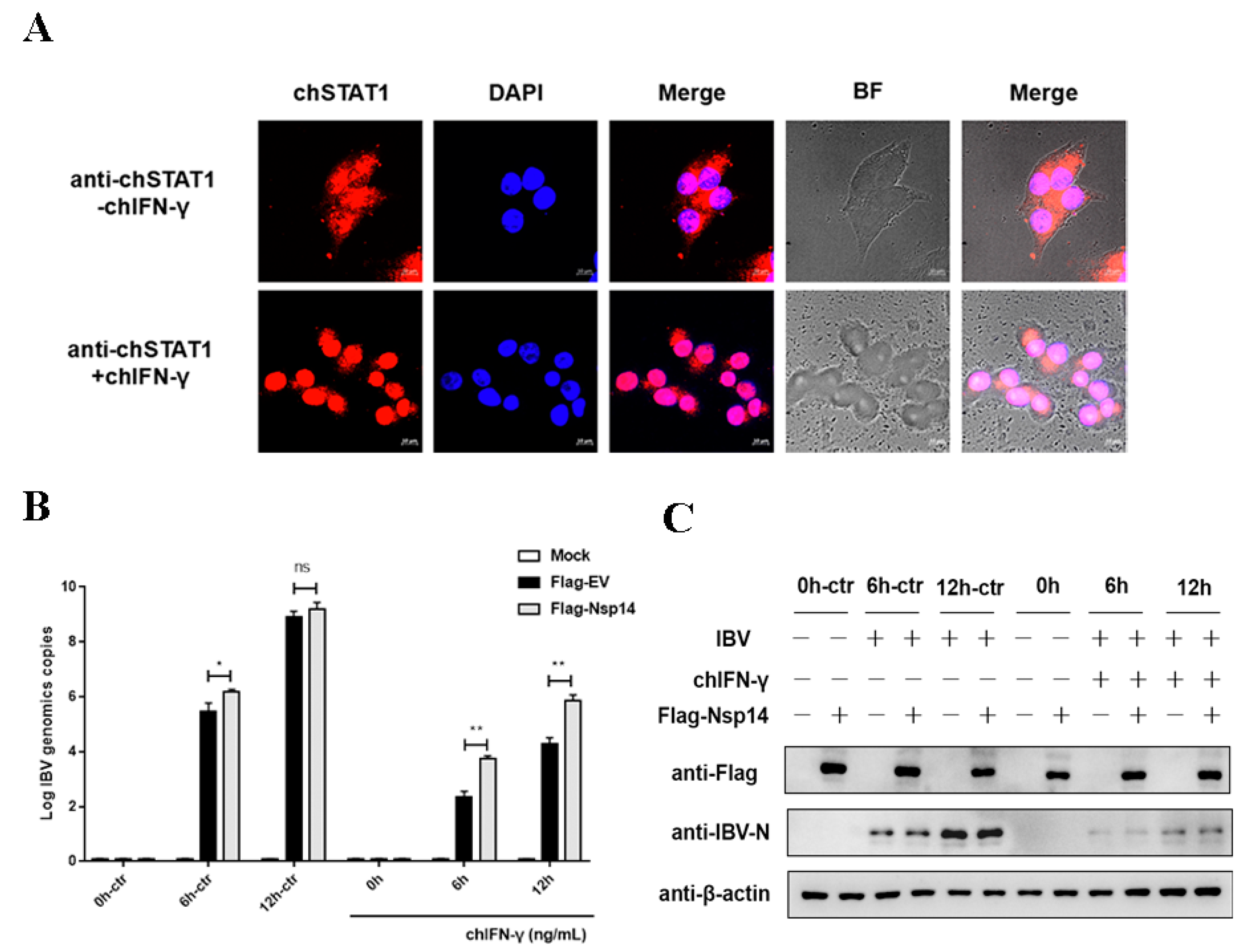
3.1. Nsp14 Protein Antagonized Type II Interferon Anti-IBV
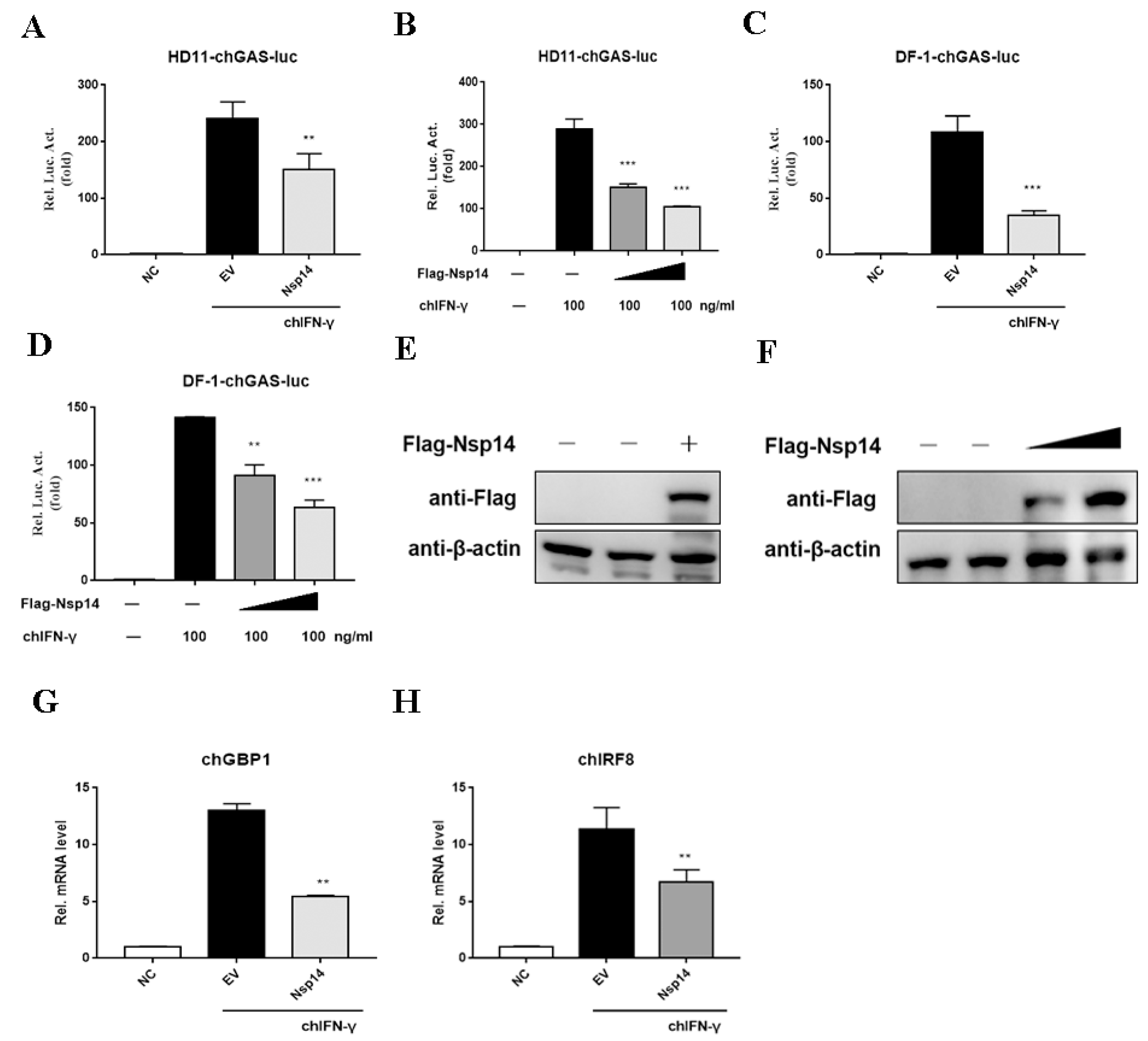
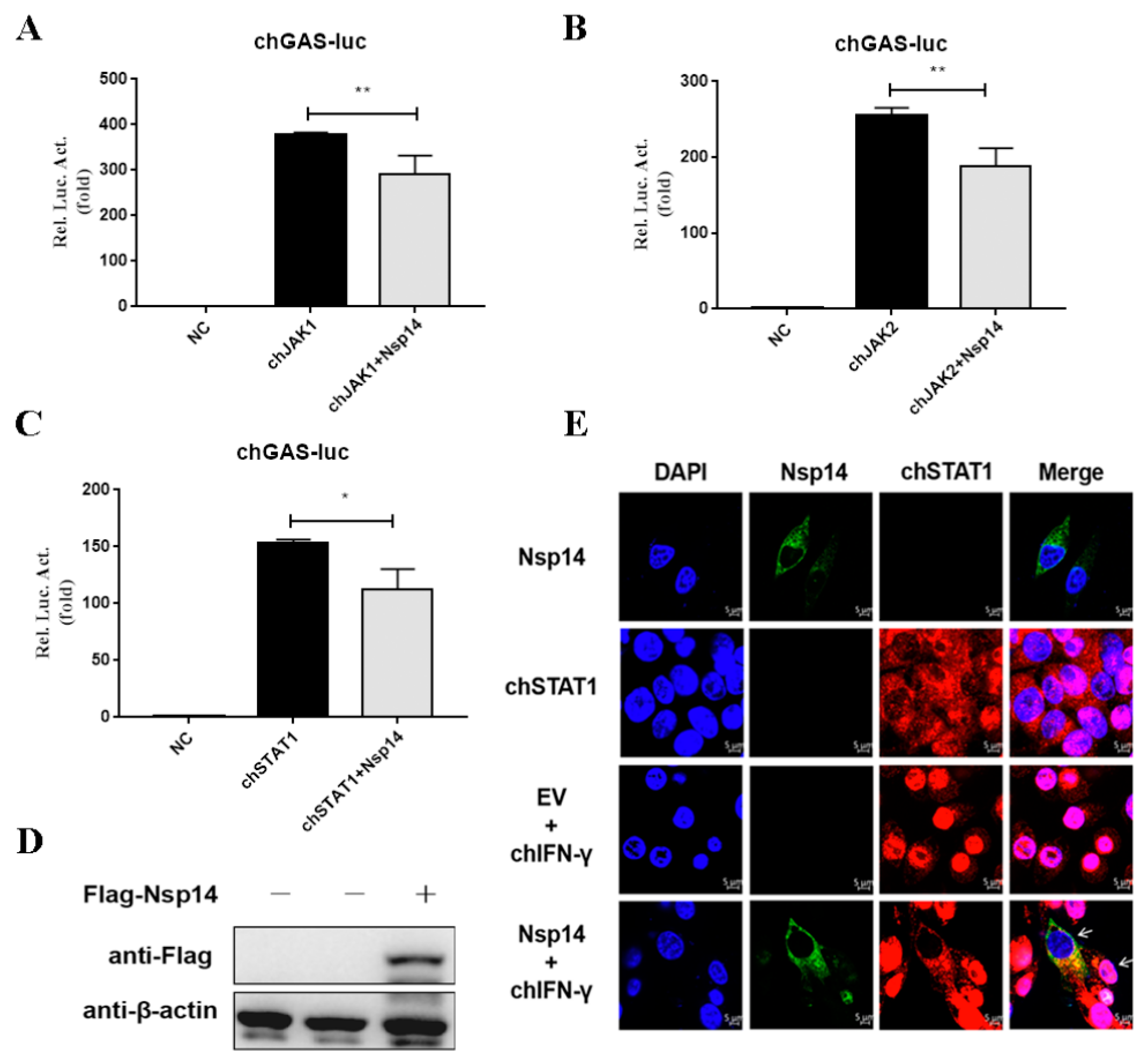
3.2. Nsp14 Protein Inhibited ChIFN-γ-Activated Signaling and Downstream Gene Expression
3.3. Nsp14 Protein Inhibited ChSTAT1 Nuclear Translocation
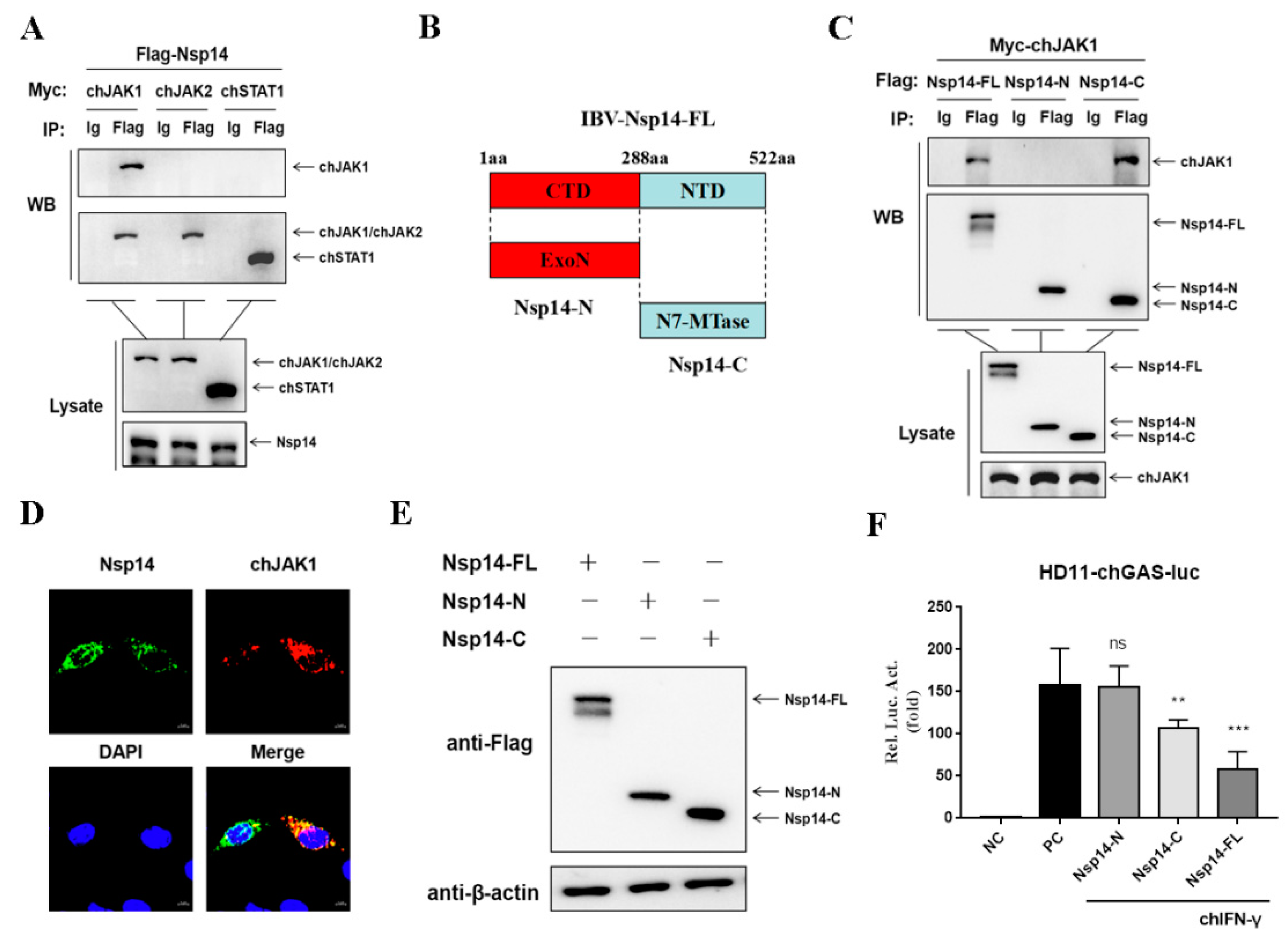
3.4. Nsp14 Protein Interacts with chJAK1 Protein
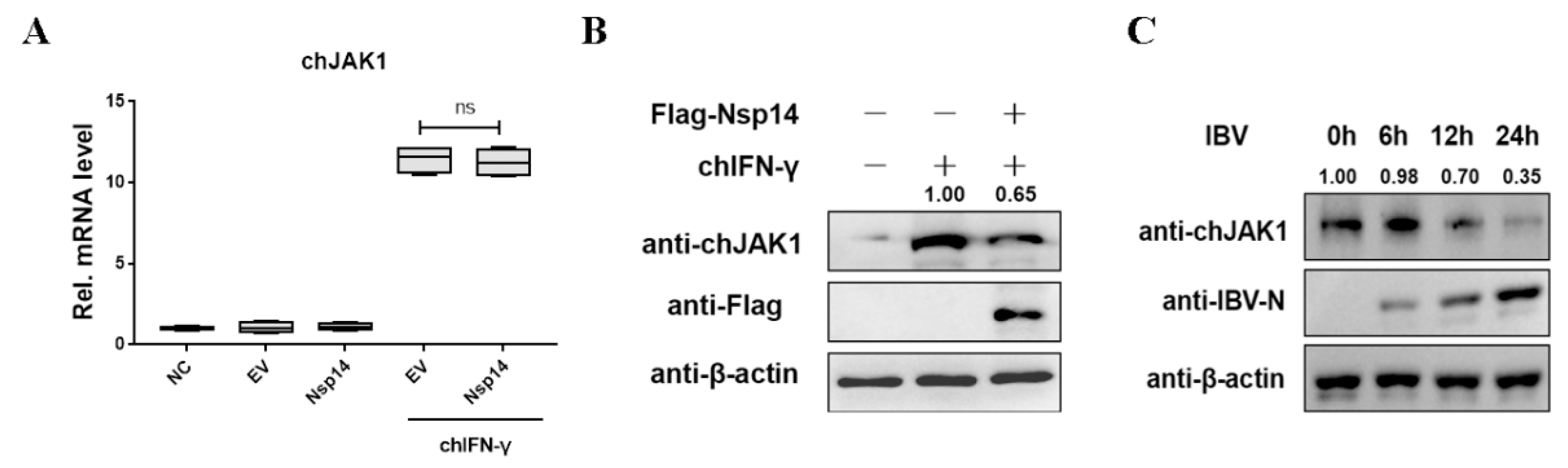
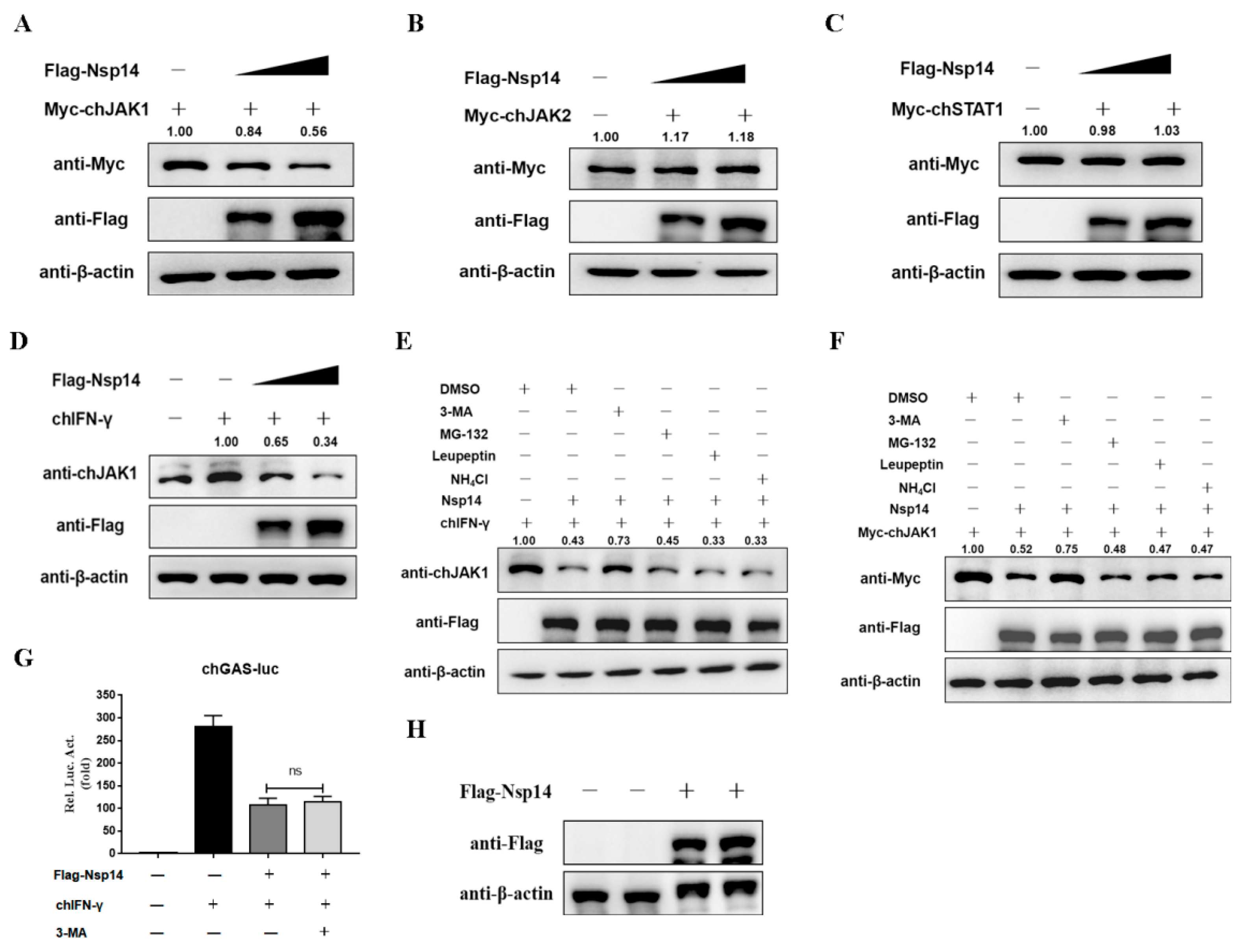
3.5. Nsp14 Protein Reduced the ChJAK1 Protein Level
3.6. Nsp14 Protein Degraded the ChJAK1 Protein via the Autophagy Pathway
4. Discussion
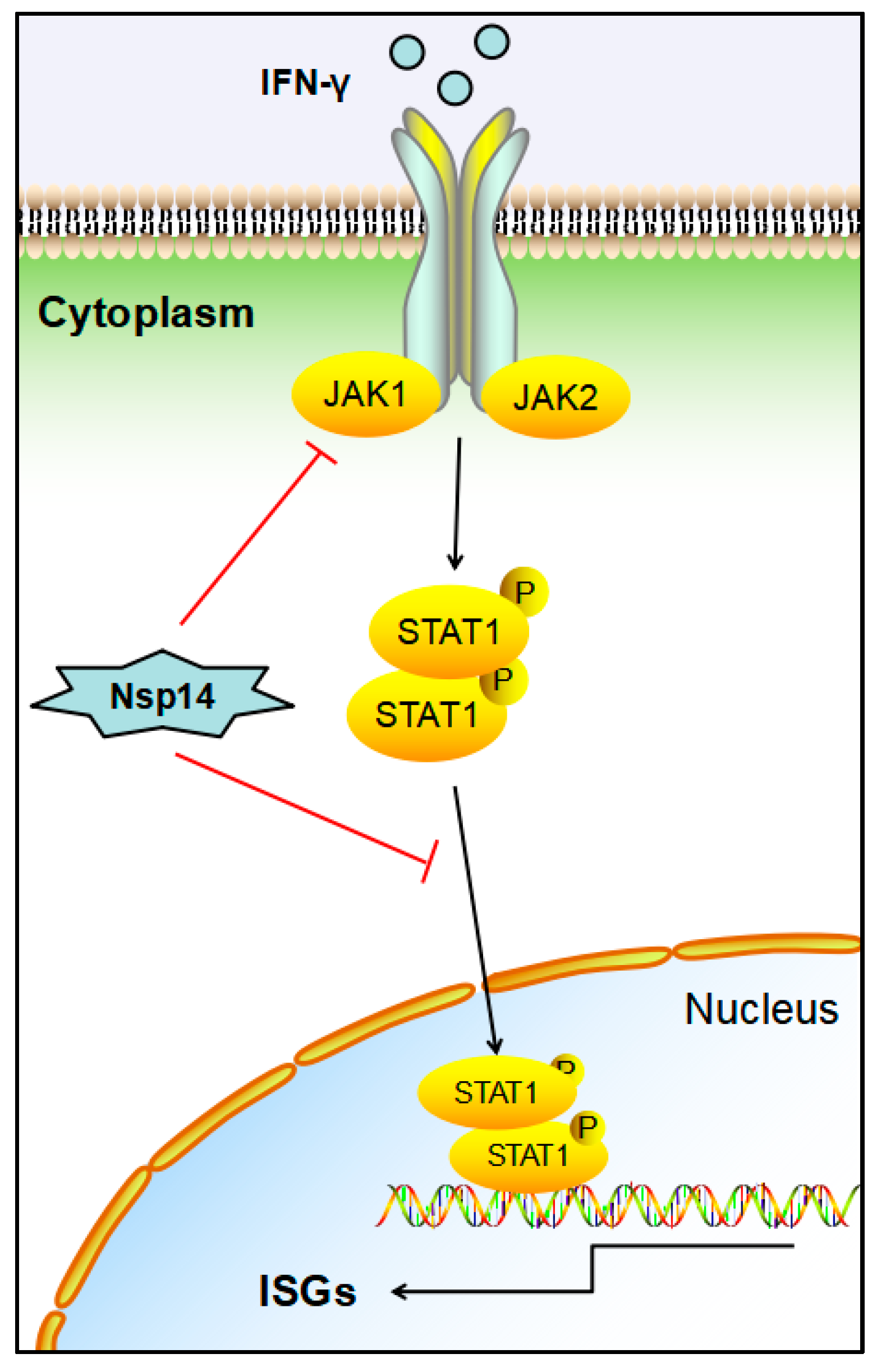
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cavanagh, D. Coronaviruses in poultry and other birds. Avian Pathol. 2005, 34, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Zhao, J.; Hu, X.; Zhang, G. Isolation and identification of four infectious bronchitis virus strains in China and analyses of their S1 glycoprotein gene. Vet. Microbiol. 2007, 122, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W. Review of infectious bronchitis virus around the world. Avian Dis. 2012, 56, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.J.; Wang, H.N.; Fan, W.Q.; Yang, X.; Zhang, A.Y.; Zeng, B.; Zhang, Y. The Nucleocapsid Protein of Avian Infectious Bronchitis Virus Interacts with Chicken Ubiquitin-Conjugating Enzyme 9 (cUBC9). J. Anim. Vet. Adv. 2012, 11, 2957–2963. [Google Scholar]
- Hosseini, A.; Hashemi, V.; Shomali, N.; Asghari, F.; Gharibi, T.; Akbari, M.; Gholizadeh, S.; Jafari, A. Innate and adaptive immune responses against coronavirus. Biomed Pharm. 2020, 132, 110859. [Google Scholar] [CrossRef]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef]
- Gribble, J.; Stevens, L.J.; Agostini, M.L.; Anderson-Daniels, J.; Chappell, J.D.; Lu, X.; Pruijssers, A.J.; Routh, A.L.; Denison, M.R. The coronavirus proofreading exoribonuclease mediates extensive viral recombination. PLoS Pathog. 2021, 17, e1009226. [Google Scholar] [CrossRef]
- Snijder, E.J.; Decroly, E.; Ziebuhr, J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. [Google Scholar]
- Shannon, A.; Le, N.T.; Selisko, B.; Eydoux, C.; Alvarez, K.; Guillemot, J.C.; Decroly, E.; Peersen, O.; Ferron, F.; Canard, B. Remdesivir and SARS-CoV-2: Structural requirements at both nsp12 RdRp and nsp14 Exonuclease active-sites. Antivir. Res. 2020, 178, 104793. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, L.; Shaw, N.; Gao, Y.; Wang, J.; Sun, Y.; Lou, Z.; Yan, L.; Zhang, R.; Rao, Z. Structural basis and functional analysis of the SARS coronavirus nsp14-nsp10 complex. Proc. Natl. Acad. Sci. USA 2015, 112, 9436–9441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, L.; Khadijah, S.; Fang, S.; Wang, L.; Tay, F.P.; Liu, D.X. The cellular RNA helicase DDX1 interacts with coronavirus nonstructural protein 14 and enhances viral replication. J. Virol. 2010, 84, 8571–8583. [Google Scholar] [CrossRef] [Green Version]
- Saramago, M.; Barria, C.; Costa, V.G.; Souza, C.S.; Viegas, S.C.; Domingues, S.; Lousa, D.; Soares, C.M.; Arraiano, C.M.; Matos, R.G. New targets for drug design: Importance of nsp14/nsp10 complex formation for the 3′–5′ exoribonucleolytic activity on SARS-CoV-2. FEBS J. 2021, 288, 5130–5147. [Google Scholar] [CrossRef] [PubMed]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [Green Version]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Devasthanam, A.S. Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence 2014, 5, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Guyer, N.B.; Severns, C.W.; Wong, P.; Feghali, C.A.; Wright, T.M. IFN-gamma induces a p91/Stat1 alpha-related transcription factor with distinct activation and binding properties. J. Immunol. 1995, 155, 3472–3480. [Google Scholar] [PubMed]
- Decker, T.; Kovarik, P.; Meinke, A. GAS elements: A few nucleotides with a major impact on cytokine-induced gene expression. J. Interferon Cytokine Res. 1997, 17, 121–134. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Aoshi, T.; Koyama, S.; Kobiyama, K.; Akira, S.; Ishii, K.J. Innate and adaptive immune responses to viral infection and vaccination. Curr. Opin. Virol. 2011, 1, 226–232. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Xu, Z.; Cao, Y. Host Antiviral Responses against Avian Infectious Bronchitis Virus (IBV): Focus on Innate Immunity. Viruses 2021, 13, 1698. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, L.; Jin, L.; Zhang, D.; Cao, X.; Guo, F.; Zhao, Y.; Bai, J.; Ma, Z.; Shang, Y.; et al. Antiviral responses of ATG13 to the infection of peste des petits ruminants virus through activation of interferon response. Gene 2020, 754, 144858. [Google Scholar] [CrossRef] [PubMed]
- Koyama, S.; Ishii, K.J.; Coban, C.; Akira, S. Innate immune response to viral infection. Cytokine 2008, 43, 336–341. [Google Scholar] [CrossRef]
- Park, A.; Iwasaki, A. Type I and Type III Interferons—Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27, 870–878. [Google Scholar] [CrossRef]
- Rose, K.M.; Elliott, R.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weiss, S.R. Murine coronavirus delays expression of a subset of interferon-stimulated genes. J. Virol. 2010, 84, 5656–5669. [Google Scholar] [CrossRef] [Green Version]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhao, J.; Perlman, S. Autocrine interferon priming in macrophages but not dendritic cells results in enhanced cytokine and chemokine production after coronavirus infection. mBio 2010, 1, 4. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Dong, X.; Ma, R.; Wang, W.; Xiao, X.; Tian, Z.; Wang, C.; Wang, Y.; Li, L.; Ren, L.; et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat. Commun. 2020, 11, 3810. [Google Scholar] [CrossRef]
- Kint, J.; Fernandez-Gutierrez, M.; Maier, H.J.; Britton, P.; Langereis, M.A.; Koumans, J.; Wiegertjes, G.F.; Forlenza, M. Activation of the chicken type I interferon response by infectious bronchitis coronavirus. J. Virol. 2015, 89, 1156–1167. [Google Scholar] [CrossRef] [Green Version]
- Roth-Cross, J.K.; Martinez-Sobrido, L.; Scott, E.P.; Garcia-Sastre, A.; Weiss, S.R. Inhibition of the alpha/beta interferon response by mouse hepatitis virus at multiple levels. J. Virol. 2007, 81, 7189–7199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeurwaerder, A.; Olyslaegers, D.A.J.; Desmarets, L.M.B.; Roukaerts, I.D.M.; Theuns, S.; Nauwynck, H.J. ORF7-encoded accessory protein 7a of feline infectious peritonitis virus as a counteragent against IFN-alpha-induced antiviral response. J. Gen. Virol. 2014, 95 Pt 2, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Treatment of SARS with human interferons. Lancet 2003, 362, 293–294. [Google Scholar] [CrossRef]
- Hart, B.J.; Dyall, J.; Postnikova, E.; Zhou, H.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Frieman, M.B.; Holbrook, M.R.; Jahrling, P.B.; et al. Interferon-beta and mycophenolic acid are potent inhibitors of Middle East respiratory syndrome coronavirus in cell-based assays. J. Gen. Virol. 2014, 95 Pt 3, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Kint, J.; Dickhout, A.; Kutter, J.; Maier, H.J.; Britton, P.; Koumans, J.; Pijlman, G.P.; Fros, J.J.; Wiegertjes, G.F.; Forlenza, M. Infectious Bronchitis Coronavirus Inhibits STAT1 Signaling and Requires Accessory Proteins for Resistance to Type I Interferon Activity. J. Virol. 2015, 89, 12047–12057. [Google Scholar] [CrossRef] [Green Version]
- Tahir, M. Coronavirus genomic nsp14-ExoN, structure, role, mechanism, and potential application as a drug target. J. Med. Virol. 2021, 93, 4258–4264. [Google Scholar] [CrossRef]
- Hayn, M.; Hirschenberger, M.; Koepke, L.; Nchioua, R.; Straub, J.H.; Klute, S.; Hunszinger, V.; Zech, F.; Prelli Bozzo, C.; Aftab, W.; et al. Systematic functional analysis of SARS-CoV-2 proteins uncovers viral innate immune antagonists and remaining vulnerabilities. Cell Rep. 2021, 35, 109126. [Google Scholar] [CrossRef]
- Moeller, N.H.; Shi, K.; Demir, O.; Belica, C.; Banerjee, S.; Yin, L.; Durfee, C.; Amaro, R.E.; Aihara, H. Structure and dynamics of SARS-CoV-2 proofreading exoribonuclease ExoN. Proc. Natl. Acad. Sci. USA 2022, 119, e2106379119. [Google Scholar] [CrossRef]
- Ogando, N.S.; El Kazzi, P.; Zevenhoven-Dobbe, J.C.; Bontes, B.W.; Decombe, A.; Posthuma, C.C.; Thiel, V.; Canard, B.; Ferron, F.; Decroly, E.; et al. Structure-function analysis of the nsp14 N7-guanine methyltransferase reveals an essential role in Betacoronavirus replication. Proc. Natl. Acad. Sci. USA 2021, 118, e2108709118. [Google Scholar] [CrossRef]
- Pan, R.; Kindler, E.; Cao, L.; Zhou, Y.; Zhang, Z.; Liu, Q.; Ebert, N.; Zust, R.; Sun, Y.; Gorbalenya, A.E.; et al. N7-Methylation of the Coronavirus RNA Cap Is Required for Maximal Virulence by Preventing Innate Immune Recognition. mBio 2022, 13, e0366221. [Google Scholar] [CrossRef]
- Case, J.B.; Li, Y.; Elliott, R.; Lu, X.; Graepel, K.W.; Sexton, N.R.; Smith, E.C.; Weiss, S.R.; Denison, M.R. Murine Hepatitis Virus nsp14 Exoribonuclease Activity Is Required for Resistance to Innate Immunity. J. Virol. 2018, 92, e01531-17. [Google Scholar] [PubMed] [Green Version]
- Hsu, J.C.; Laurent-Rolle, M.; Pawlak, J.B.; Wilen, C.B.; Cresswell, P. Translational shutdown and evasion of the innate immune response by SARS-CoV-2 NSP14 protein. Proc. Natl. Acad. Sci. USA 2021, 118, e2101161118. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Luo, Y.; O’Shea, J.J.; Nakayamada, S. Janus kinase-targeting therapies in rheumatology: A mechanisms-based approach. Nat. Rev. Rheumatol. 2022, 18, 133–145. [Google Scholar] [PubMed]
- Ezeonwumelu, I.J.; Garcia-Vidal, E.; Ballana, E. JAK-STAT Pathway: A Novel Target to Tackle Viral Infections. Viruses 2021, 13, 2379. [Google Scholar]
- Meyer, T.; Vinkemeier, U. Nucleocytoplasmic shuttling of STAT transcription factors. Eur. J. Biochem. 2004, 271, 4606–4612. [Google Scholar] [CrossRef]
- Frieman, M.; Yount, B.; Heise, M.; Kopecky-Bromberg, S.A.; Palese, P.; Baric, R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007, 81, 9812–9824. [Google Scholar] [PubMed] [Green Version]
- Gu, W.; Gan, H.; Ma, Y.; Xu, L.; Cheng, Z.J.; Li, B.; Zhang, X.; Jiang, W.; Sun, J.; Sun, B.; et al. The molecular mechanism of SARS-CoV-2 evading host antiviral innate immunity. Virol. J. 2022, 19, 49. [Google Scholar]
- Fung, S.Y.; Siu, K.L.; Lin, H.; Chan, C.P.; Yeung, M.L.; Jin, D.Y. SARS-CoV-2 NSP13 helicase suppresses interferon signaling by perturbing JAK1 phosphorylation of STAT1. Cell Biosci. 2022, 12, 36. [Google Scholar] [CrossRef]
- Feng, K.; Min, Y.Q.; Sun, X.; Deng, F.; Li, P.; Wang, H.; Ning, Y.J. Interactome profiling reveals interaction of SARS-CoV-2 NSP13 with host factor STAT1 to suppress interferon signaling. J. Mol. Cell Biol. 2021, 13, 760–762. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Forward (5′–3′) | Reverse (5′–3′) |
|---|---|---|
| IBV-N | GAAGAAAACCAGTCCCAGA | TTACCAGCAACCCACAC |
| GAPDH | CATCACAGCCACACAGAAG | GGTCAGGTCAACAACAGAGA |
| chJAK1 | AGATGATGAGAATGAAGGATA | ACGATGTGCTTATGAGAA |
| chGBP1 | AAGTCCTTCCTGATGAACC | CTTGGTCTCCGCATACAC |
| chIRF8 | CAGGACTACAACCAGGAG | ACCTTCTTTGAATTTGCCTTT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, P.; Gu, K.; Li, H.; Zhao, Y.; Li, C.; Wen, R.; Zhou, C.; Lei, C.; Yang, X.; Wang, H. Infectious Bronchitis Virus Nsp14 Degrades JAK1 to Inhibit the JAK-STAT Signaling Pathway in HD11 Cells. Viruses 2022, 14, 1045. https://doi.org/10.3390/v14051045
Ma P, Gu K, Li H, Zhao Y, Li C, Wen R, Zhou C, Lei C, Yang X, Wang H. Infectious Bronchitis Virus Nsp14 Degrades JAK1 to Inhibit the JAK-STAT Signaling Pathway in HD11 Cells. Viruses. 2022; 14(5):1045. https://doi.org/10.3390/v14051045
Chicago/Turabian StyleMa, Peng, Kui Gu, Hao Li, Yu Zhao, Chao Li, Renqiao Wen, Changyu Zhou, Changwei Lei, Xin Yang, and Hongning Wang. 2022. "Infectious Bronchitis Virus Nsp14 Degrades JAK1 to Inhibit the JAK-STAT Signaling Pathway in HD11 Cells" Viruses 14, no. 5: 1045. https://doi.org/10.3390/v14051045