
Molecular Characterization of Outer Capsid Proteins VP5 and VP7 of Grass Carp Reovirus


Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells, Virus, and Antibodies
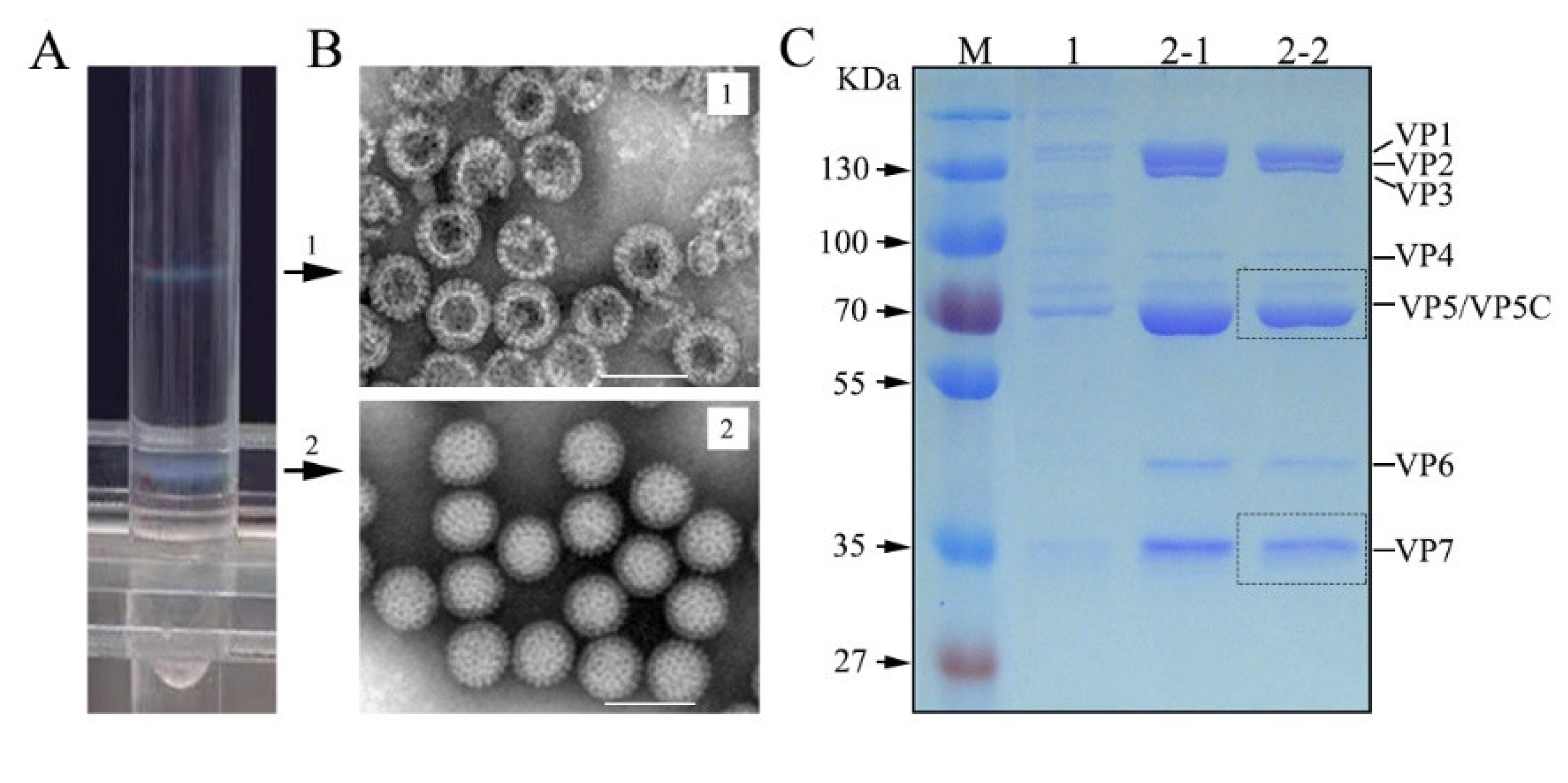
2.2. Virus Particle Purification
2.3. Generation of Recombinant Baculoviruses to Express VP7 and VP5/VP5N42A
2.4. In Vitro Expression of VP5 and VP7 Proteins
2.5. SDS-PAGE and Immunoblotting
2.6. Immunofluorescence and Fluorescent Focus Assay
2.7. Trypsin Digestion, LC-MS/MS Analysis, and Data Analyses
2.8. Limited Early Digestion of Virions with Trypsin
2.9. Transmission Electron Microscopy and Cryo-EM Image Preparation
3. Results
3.1. GCRV Purification and Structural Protein Analysis
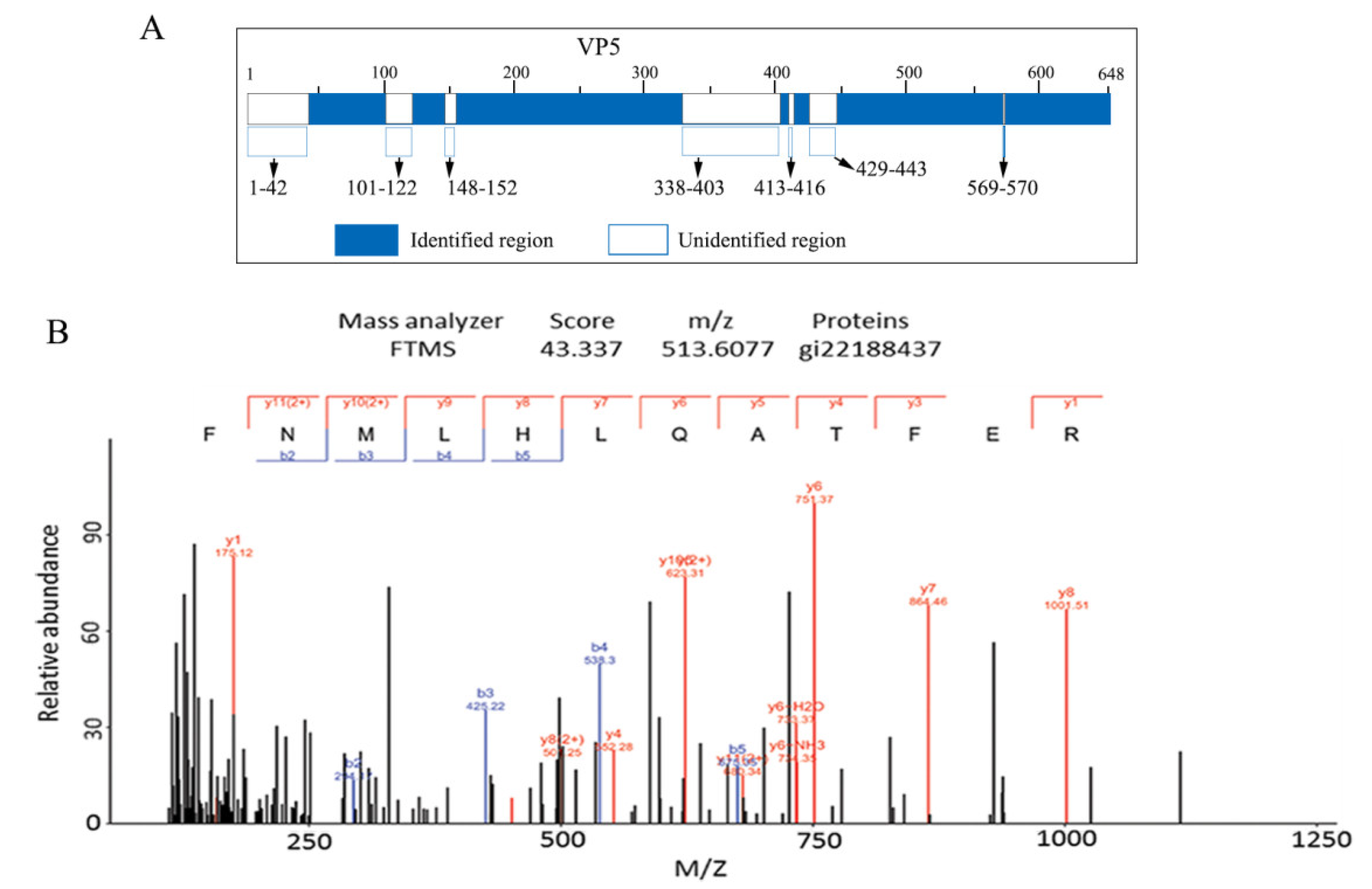
3.2. MS Assays of GCRV OCP VP5
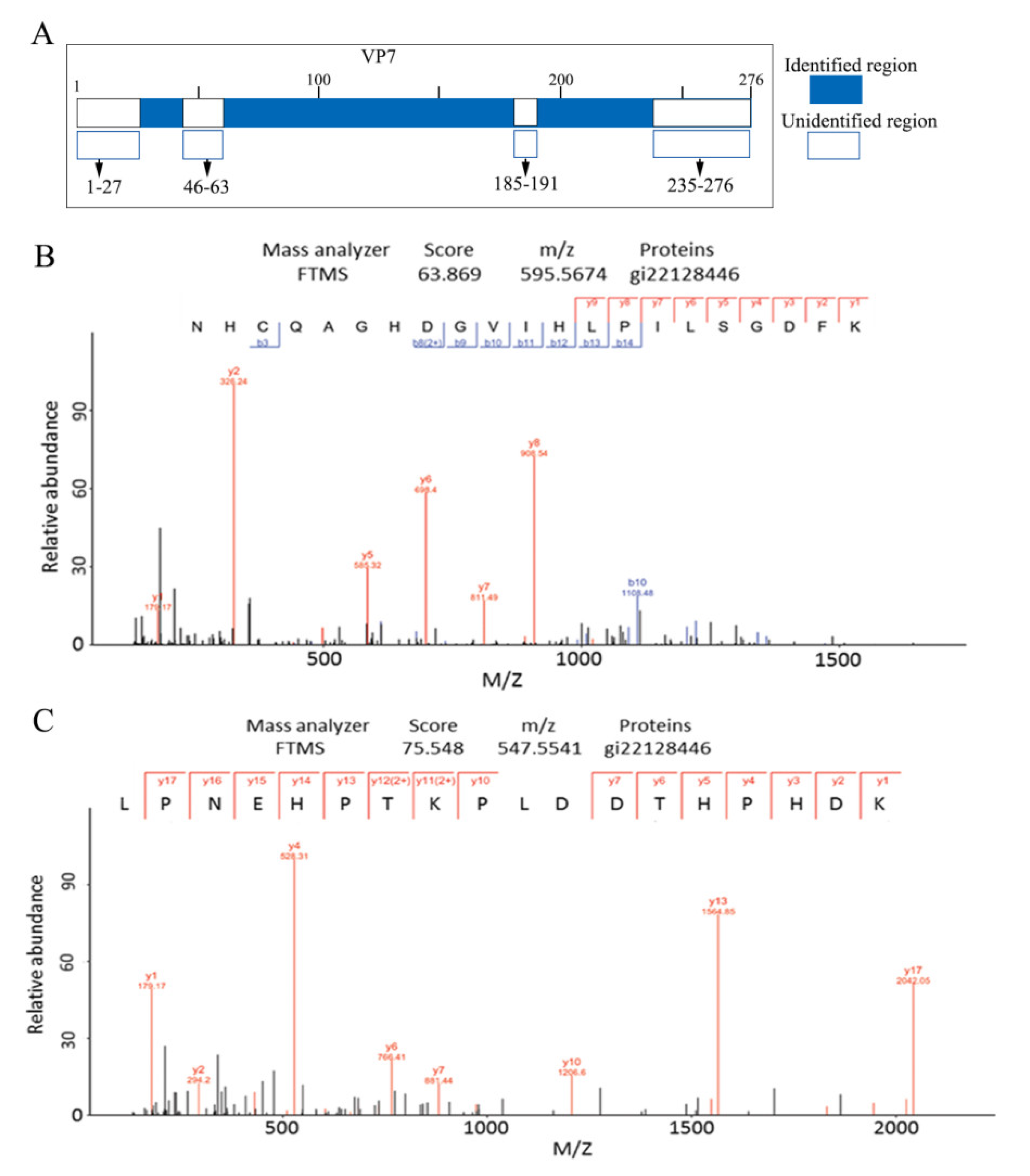
3.3. MS Assays of the GCRV Outermost Capsid Protein VP7
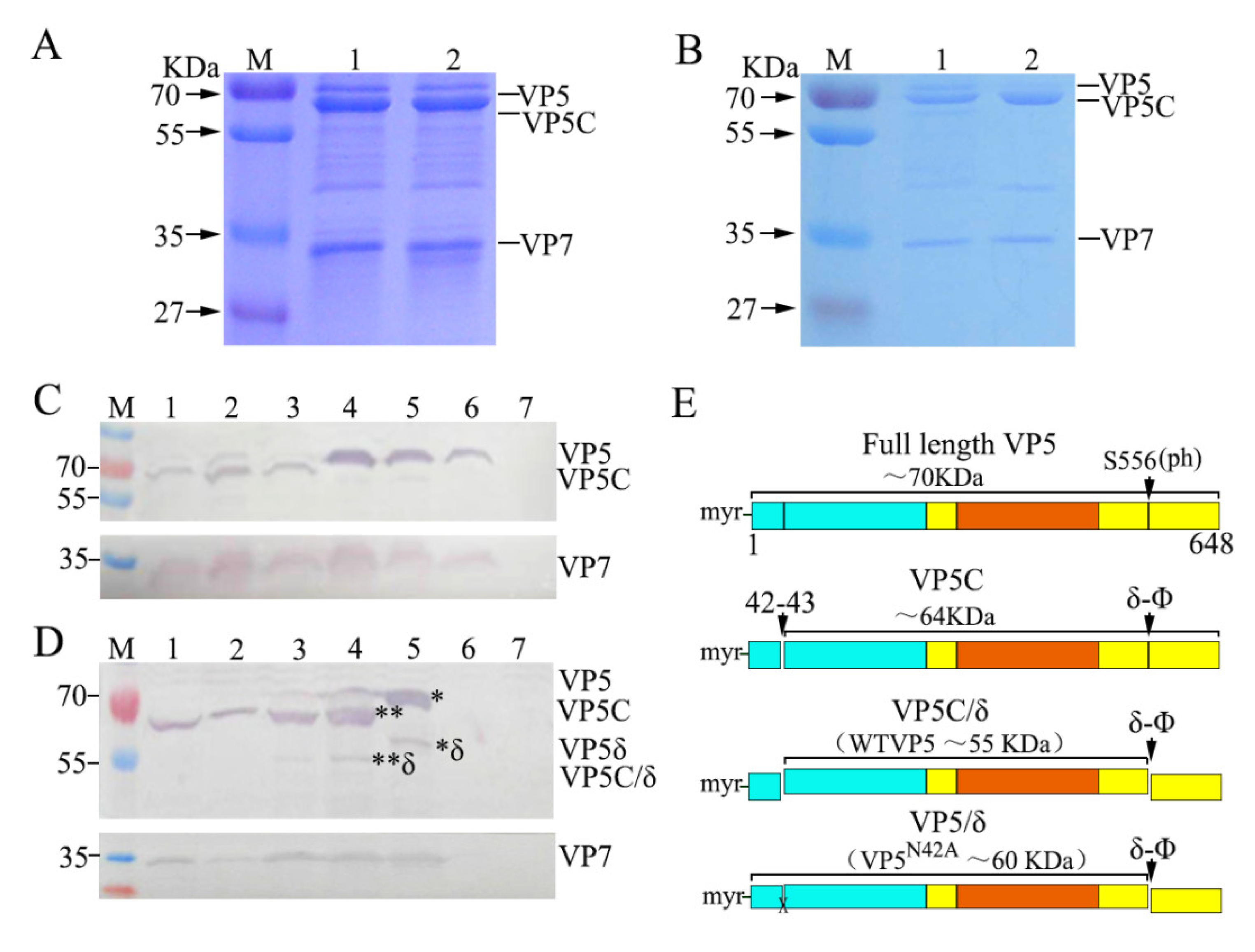
3.4. Autocleavage of VP5 between Residues Asn42 and Pro43 Detected in Native Particles and Lysates of Infected Cells
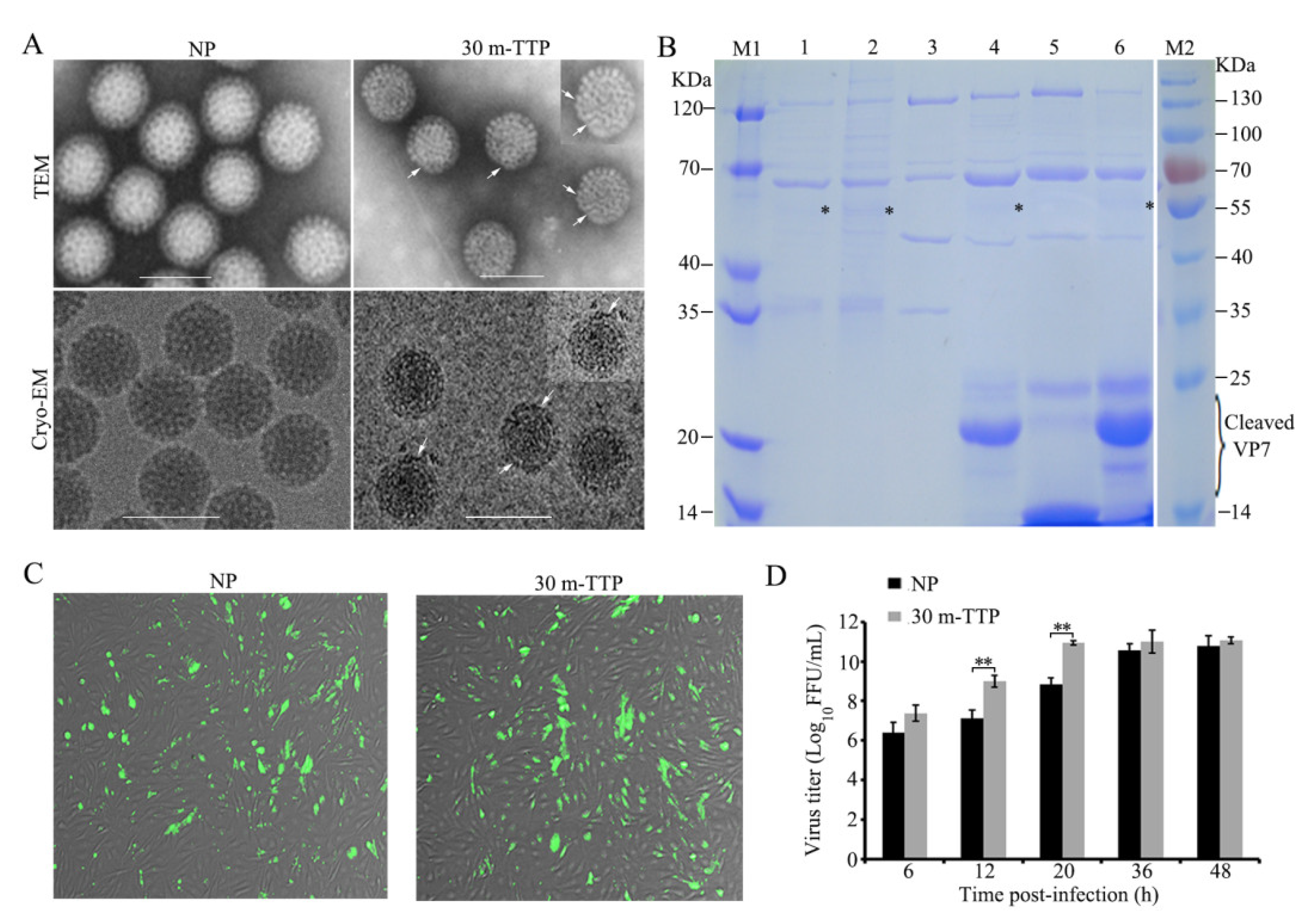
3.5. Cleavage Fragments of VP7 with Enhanced Infectivity were Detected with Limited Early Tryptic Digestion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kim, J.; Tao, Y.; Reinisch, K.M.; Harrison, S.C.; Nibert, M.L. Orthoreovirus and Aquareovirus core proteins: Conserved enzymatic surfaces, but not protein-protein interfaces. Virus Res. 2004, 101, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Liemann, S.; Chandran, K.; Baker, T.S.; Nibert, M.L.; Harrison, S.C. Structure of the reovirus membrane-penetration protein, μ1, in a complex with its protector protein, σ3. Cell 2002, 108, 283–295. [Google Scholar] [CrossRef] [Green Version]
- Odegard, A.L.; Chandran, K.; Zhang, X.; Parker, J.S.; Baker, T.S.; Nibert, M.L. Putative autocleavage of outer capsid protein μ1, allowing release of myristoylated peptide μ1N during particle uncoating, is critical for cell entry by reovirus. J. Virol. 2004, 78, 8732–8745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillotson, L.; Shatkin, A.J. Reovirus polypeptide sigma 3 and N-terminal myristoylation of polypeptide mu 1 are required for site-specific cleavage to mu 1C in transfected cells. J. Virol. 1992, 66, 2180–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gummersheimer, S.L.; Snyder, A.J.; Danthi, P. Control of Capsid Transformations during Reovirus Entry. Viruses 2021, 13, 153. [Google Scholar] [CrossRef]
- Drayna, D.; Fields, B.N. Activation and characterization of the reovirus transcriptase: Genetic analysis. J. Virol. 1982, 41, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Knipe, D.M.; Howley, P.M.; Griffin, D.E.; Lamb, R.A.; Martin, M.A.; Roizman, B.; Straus, S.E. Fields Virology, 6th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1304–1613. Available online: https://www.wolterskluwer.com/en/solutions/ovid/fields-virology-6th-edition-15514 (accessed on 9 May 2022).
- King, A.M.; Lefkowitz, E.; Adams, M.J.; Carstens, E.B. Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses, 1st ed.; Elsevier: London, UK, 2012; pp. 541–637. Available online: https://doi.org/10.1016/B978-0-12-384684-6.00051-3 (accessed on 9 May 2022).
- Fang, Q.; Zhang, J.; Zhang, F.; Lv, L.; Zhang, K.; Ma, J.; Fan, Y.; Zeng, W.; Wang, Y.; Wang, Q.; et al. Aquareovirus, 1st ed.; Springer: Singapore, 2021; Available online: https://link.springer.com/book/10.1007/978-981-16-1903-8 (accessed on 9 May 2022).
- Skoge, R.H.; Nylund, A.; Solheim, K.; Plarre, H. Complete genome of Atlantic halibut reovirus (AHRV) associated with mortality in production of Atlantic halibut (Hippoglossus hippoglossus) fry. Aquaculture 2019, 509, 23–31. [Google Scholar] [CrossRef]
- Subramanian, K.; McPhillips, T.H.; Samal, S.K. Characterization of the polypeptides and determination of genome coding assignments of an aquareovirus. Virology 1994, 205, 75–81. [Google Scholar] [CrossRef]
- Winton, J.; Lannan, C.; Fryer, J.; Hedrick, R.; Meyers, T.; Plumb, J.; Yamamoto, T. Morphological and biochemical properties of four members of a novel group of reoviruses isolated from aquatic animals. J. Gen. Virol. 1987, 68, 353–364. [Google Scholar] [CrossRef]
- Jaafar, F.M.; Goodwin, A.E.; Belhouchet, M.; Merry, G.; Fang, Q.; Cantaloube, J.F.; Biagini, P.; de Micco, P.; Mertens, P.P.; Attoui, H. Complete characterisation of the American grass carp reovirus genome (genus Aquareovirus: Family Reoviridae) reveals an evolutionary link between aquareoviruses and coltiviruses. Virology 2008, 373, 310–321. [Google Scholar] [CrossRef] [Green Version]
- Rangel, A.A.; Rockemann, D.D.; Hetrick, F.M.; Samal, S.K. Identification of grass carp haemorrhage virus as a new genogroup of aquareovirus. J. Gen. Virol. 1999, 80, 2399–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, W.; Bergmannc, S.M.; Dong, H.; Yang, Y.; Wu, M.; Liu, H.; Chen, Y.; Li, H. Identification, virulence, and molecular characterization of a recombinant isolate of grass carp reovirus genotype I. Viruses 2021, 13, 807. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Liu, W.; Li, Y.; Zhou, Y.; Meng, Y.; Zeng, L.; Vakharia, V.N.; Fan, Y. Isolation, identification, and genomic analysis of a novel reovirus from healthy grass carp and its dynamic proliferation in vitro and in vivo. Viruses 2021, 13, 690. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Rao, S.; Zeng, L.; Ma, J.; Zhou, Y.; Xu, J.; Zhang, H. Identification and genomic characterization of a novel fish reovirus, Hubei grass carp disease reovirus, isolated in 2009 in China. J. Gen. Virol. 2013, 94, 2266–2277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, C.; Ke, F.; Chen, Z.; Zhang, Q. Complete genome sequence and comparative analysis of grass carp reovirus strain 109 (GCReV-109) with other grass carp reovirus strains reveals no significant correlation with regional distribution. Arch. Virol. 2014, 159, 2435–2440. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zeng, W.; Liu, C.; Zhang, C.; Wang, Y.; Shi, C.; Wu, S. Complete genome sequence of a reovirus isolated from grass carp, indicating different genotypes of GCRV in China. J. Virol. 2012, 86, 12466. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Tian, Y.; Deng, G.; Chi, Y.; Jiang, X. Complete genomic sequence of a reovirus isolated from grass carp in China. Virus Res. 2012, 163, 275–283. [Google Scholar] [CrossRef]
- Nibert, M.L.; Duncan, R. Bioinformatics of recent aqua-and orthoreovirus isolates from fish: Evolutionary gain or loss of FAST and fiber proteins and taxonomic implications. PLoS ONE 2013, 8, e68607. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Fang, Q.; Shah, S.; Atanasov, I.C.; Zhou, Z.H. Subnanometer-resolution structures of the grass carp reovirus core and virion. J. Mol. Biol. 2008, 382, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Shah, S.; Liang, Y.; Zhou, H. 3D reconstruction and capsid protein characterization of grass carp reovirus. Sci. in China Series C: Life Sci. 2005, 48, 593–600. [Google Scholar] [CrossRef]
- Shaw, A.L.; Samal, S.K.; Subramanian, K.; Prasad, B.V. The structure of aquareovirus shows how the different geometries of the two layers of the capsid are reconciled to provide symmetrical interactions and stabilization. Structure 1996, 4, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, F.; Su, R.; Li, X.; Chen, W.; Chen, Q.; Yang, T.; Wang, J.; Liu, H.; Fang, Q. Structure of RNA polymerase complex and genome within a dsRNA virus provides insights into the mechanisms of transcription and assembly. Proc. Natl. Acad. Sci. USA 2018, 115, 7344–7349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Jin, L.; Fang, Q.; Hui, W.H.; Zhou, Z. 3.3 Å cryo-EM structure of a nonenveloped virus reveals a priming mechanism for cell entry. Cell 2010, 141, 472–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, X.; Parent, K.N.; Goodman, R.P.; Tang, J.; Shou, J.; Nibert, M.L.; Duncan, R.; Baker, T.S. Virion structure of baboon reovirus, a fusogenic orthoreovirus that lacks an adhesion fiber. J. Virol. 2011, 85, 7483–7495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPhillips, T.H.; Dinan, D.; Subramanian, K.; Samal, S.K. Enhancement of aquareovirus infectivity by treatment with proteases: Mechanism of action. J. Virol. 1998, 72, 3387–3389. [Google Scholar] [CrossRef] [Green Version]
- Fang, Q.; Seng, E.; Ding, Q.; Zhang, L. Characterization of infectious particles of grass carp reovirus by treatment with proteases. Arch. Virol. 2008, 153, 675–682. [Google Scholar] [CrossRef]
- Guo, H.; Sun, X.; Yan, L.; Shao, L.; Fang, Q. The NS16 protein of aquareovirus-C is a fusion-associated small transmembrane (FAST) protein, and its activity can be enhanced by the nonstructural protein NS26. Virus Res. 2013, 171, 129–137. [Google Scholar] [CrossRef]
- Guo, H.; Chen, Q.; Yan, L.; Zhang, J.; Yan, S.; Zhang, F.; Fang, Q. Identification of a functional motif in the AqRV NS26 protein required for enhancing the fusogenic activity of FAST protein NS16. J. Gen. Virol. 2015, 96, 1080–1085. [Google Scholar] [CrossRef]
- Shao, L.; Guo, H.; Yan, L.M.; Liu, H.; Fang, Q. Aquareovirus NS80 recruits viral proteins to its inclusions, and its C-terminal domain is the primary driving force for viral inclusion formation. PLoS ONE 2013, 8, e55334. [Google Scholar] [CrossRef]
- Yu, F.; Wang, L.; Li, W.; Lu, L. Identification of a novel membrane-associated protein from the S7 segment of grass carp reovirus. J. Gen. Virol. 2019, 100, 369–379. [Google Scholar] [CrossRef]
- Cheng, L.; Zhu, J.; Hui, W.H.; Zhang, X.; Honig, B.; Fang, Q.; Zhou, Z. Backbone model of an aquareovirus virion by cryo-electron microscopy and bioinformatics. J. Mol. Biol. 2010, 397, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Shao, L.; Sun, X.; Fang, Q. Antibodies against outer-capsid proteins of grass carp reovirus expressed in E. coli are capable of neutralizing viral infectivity. Virol. J. 2011, 8, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.E.; Zweerink, H.J.; Joklik, W.K. Polypeptide components of virions, top component and cores of reovirus type 3. Virology 1969, 39, 791–810. [Google Scholar] [CrossRef]
- Yan, S.; Zhang, J.; Guo, H.; Yan, L.; Chen, Q.; Zhang, F.; Fang, Q. VP5 autocleavage is required for efficient infection by in vitro-recoated aquareovirus particles. J. Gen. Virol. 2015, 96, 1795–1800. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, H.; Li, X.; Fang, Q. The VP2 protein of grass carp reovirus (GCRV) expressed in a baculovirus exhibits RNA polymerase activity. Virol. Sin. 2014, 29, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Seng, E.K.; Dai, W.; Zhang, L. Construction and co-expression of grass carp reovirus VP6 protein and enhanced green fluorescence protein in the insect cells. Virol. Sin. 2007, 22, 397–404. [Google Scholar] [CrossRef]
- Zhang, F.; Guo, H.; Zhang, J.; Chen, Q.; Fang, Q. Identification of the caveolae/raft-mediated endocytosis as the primary entry pathway for aquareovirus. Virology 2018, 513, 195–207. [Google Scholar] [CrossRef]
- Guo, H.; Zhang, J.; Wang, Y.; Bu, C.; Zhou, Y.; Fang, Q. Comparative proteomic analysis of lysine acetylation in fish CIK cells infected with aquareovirus. Int. J. Mol. Sci. 2017, 18, 2419. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Cheng, Z.; Zhu, J.; Xu, W.; Peng, X.; Chen, C.; Li, W.; Wang, F.; Cao, L.; Yi, X. Suberoylanilide hydroxamic acid treatment reveals crosstalks among proteome, ubiquitylome and acetylome in non-small cell lung cancer A549 cell line. Sci. Rep. 2015, 5, 9520. [Google Scholar] [CrossRef] [Green Version]
- Attoui, H.; Fang, Q.; Jaafar, F.M.; Cantaloube, J.F.; Biagini, P.; de Micco, P.; de Lamballerie, X. Common evolutionary origin of aquareoviruses and orthoreoviruses revealed by genome characterization of Golden shiner reovirus, Grass carp reovirus, Striped bass reovirus and golden ide reovirus (genus Aquareovirus, family Reoviridae). J. Gen. Virol. 2002, 83, 1941–1951. [Google Scholar] [CrossRef] [PubMed]
- Dryden, K.A.; Wang, G.; Yeager, M.; Nibert, M.L.; Coombs, K.M.; Furlong, D.B.; Fields, B.N.; Baker, T.S. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: Analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J. Cell. Biol. 1993, 122, 1023–1041. [Google Scholar] [CrossRef] [PubMed]
- Nibert, M.L.; Odegard, A.L.; Agosto, M.A.; Chandran, K.; Schiff, L.A. Putative autocleavage of reovirus μ1 protein in concert with outer-capsid disassembly and activation for membrane permeabilization. J. Mol. Biol. 2005, 345, 461–474. [Google Scholar] [CrossRef] [PubMed]
- Modrof, J.; Lymperopoulos, K.; Roy, P. Phosphorylation of bluetongue virus nonstructural protein 2 is essential for formation of viral inclusion bodies. J. Virol. 2005, 79, 10023–10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Law, L.M.J.; Everitt, J.C.; Beatch, M.D.; Holmes, C.F.; Hobman, T.C. Phosphorylation of rubella virus capsid regulates its RNA binding activity and virus replication. J. Virol. 2003, 77, 1764–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilia, M.; Johnson, R.; Sweeney, M.; DeBlasio, S.L.; Bruce, J.E.; MacCoss, M.J.; Gray, S.M. Evidence for lysine acetylation in the coat protein of a polerovirus. J. Gen. Virol. 2014, 95, 2321. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Fung, S.; Xie, G.; Wong, L.; Jin, D.; Cai, Z. Identification of lysine acetylation sites on MERS-CoV replicase pp1ab. Mol. Cell Proteom. 2020, 19, 1303–1309. [Google Scholar] [CrossRef]
- Gummersheimer, S.L.; Danthi, P. Reovirus core proteins λ1 and σ2 promote stability of disassembly intermediates and influence early replication events. J. Virol. 2020, 94, e00491-20. [Google Scholar] [CrossRef]
- Glover, K.K.; Sutherland, D.M.; Dermody, T.S.; Coombs, K.M. A single point mutation, Asn16→Lys, dictates the temperature-sensitivity of the reovirus tsG453 mutant. Viruses 2021, 13, 289. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Fragment# | Start-End | Peptide Sequence | Type of Modification/Site | Mass Charge Ratio (m/z) |
|---|---|---|---|---|
| 1 1 | 162–184 162–184 | HLDTAMTMLTPDISAGSASCfigNWK HLDTAMTMLTPDISAGSASCNWK | Phosphorylation/Thr165 Phosphorylation/Thr171 | 2181.5347, 2+ 849.3580, 3+ |
| 2 | 215–226 | YPALKPGNPDTK | Acetylation/Lys219 | 671.8549, 2+ |
| 3 | 270–292 | DLDLIEADTPLPVSVFTPSLAPR | Phosphorylation/ser283 | 849.4308, 3+ |
| 4 | 549–567 | LSQVGQASPTPPDYPLLWR | Phosphorylation/Ser556 | 1103.0388, 2+ |
| 5 | 616–625 | GVTDASEKLR | Acetylation/Lys623 | 559.2952, 2+ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, F.; Sun, D.; Fang, Q. Molecular Characterization of Outer Capsid Proteins VP5 and VP7 of Grass Carp Reovirus. Viruses 2022, 14, 1032. https://doi.org/10.3390/v14051032
Zhang F, Sun D, Fang Q. Molecular Characterization of Outer Capsid Proteins VP5 and VP7 of Grass Carp Reovirus. Viruses. 2022; 14(5):1032. https://doi.org/10.3390/v14051032
Chicago/Turabian StyleZhang, Fuxian, Diangang Sun, and Qin Fang. 2022. "Molecular Characterization of Outer Capsid Proteins VP5 and VP7 of Grass Carp Reovirus" Viruses 14, no. 5: 1032. https://doi.org/10.3390/v14051032