
Oncolytic H-1 Parvovirus Hijacks Galectin-1 to Enter Cancer Cells

 ,
,
Abstract
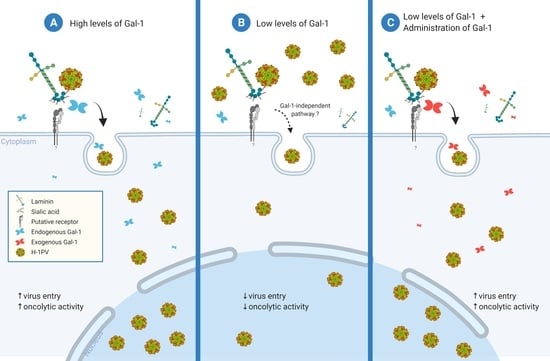
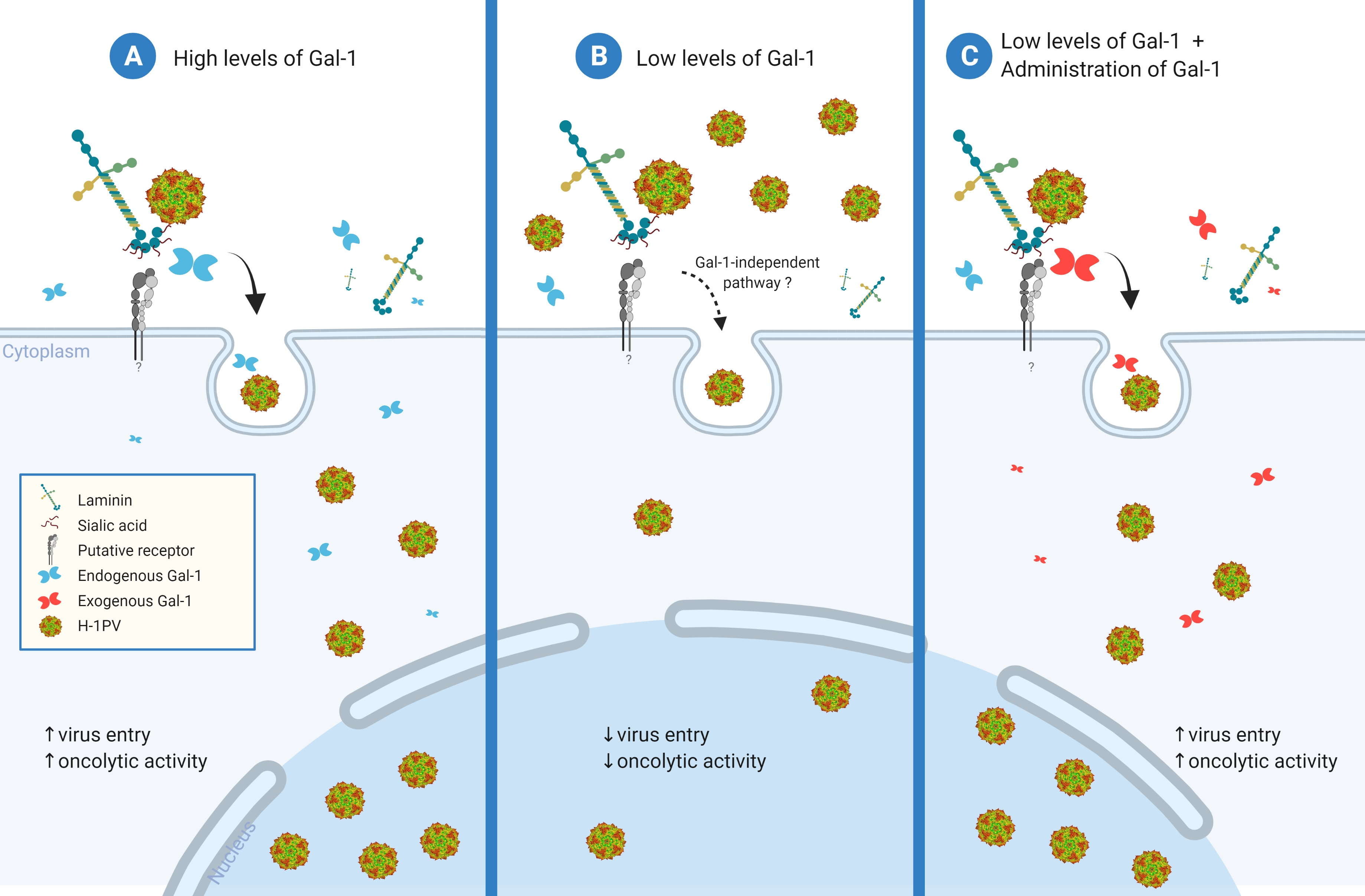
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Viruses
2.3. siRNA-Mediated Knockdown
2.4. Viral Transduction Assay
2.5. Fluorescence Microscopy
2.6. Lactose Pre-treatment for H-1PV Transduction Analysis
2.7. Western Blotting
2.8. Generation of the LGALS1 Knockout Cell Line
2.9. Plasmid Transfection
2.10. Confocal Microscopy
2.11. MTT Viability Assay
2.12. Binding-Only and Binding and Entry Assays
2.13. Flow Cytometry
2.14. Tissue Microarray
2.15. Correlation Analysis between Gene Expression of Cancer Cell Lines and H-1PV-Induced Oncolysis
2.16. Measurement of Transcript Levels
2.17. xCELLigence
2.18. Statistical Analysis
3. Results
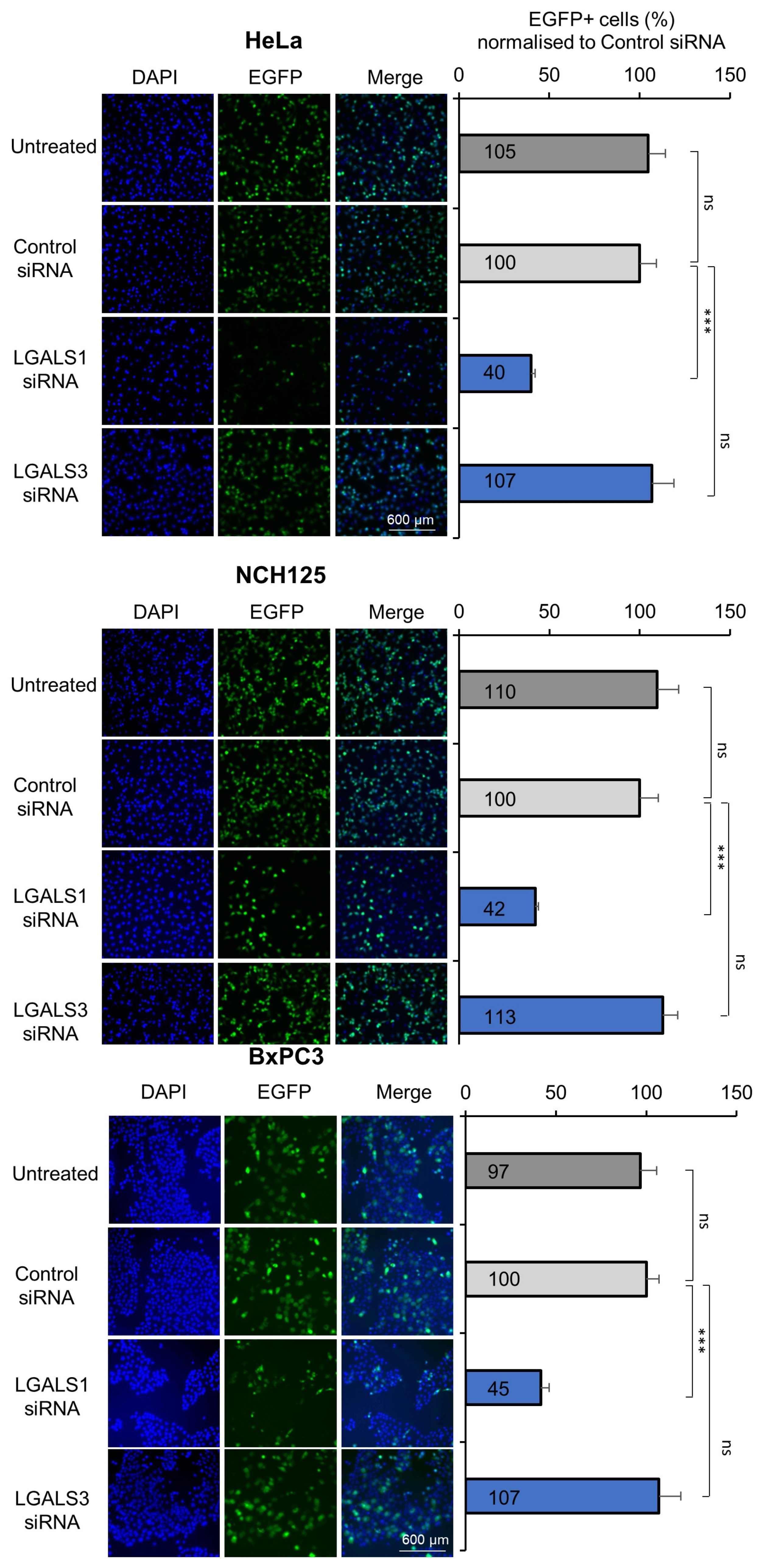
3.1. Knockdown of LGALS1, but Not LGALS3, Hampers H-1PV Infection
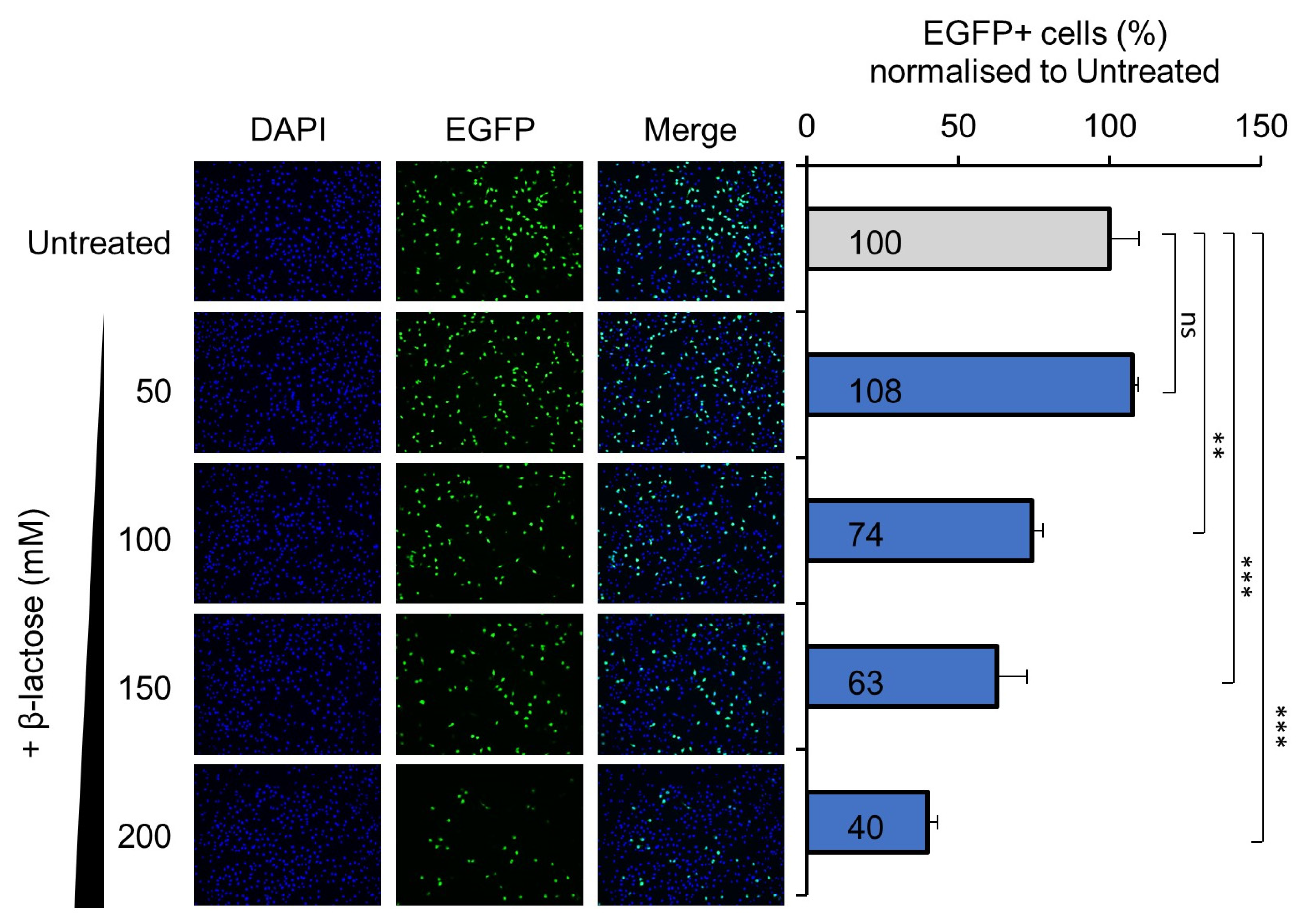
3.2. Pre-Treatment with Lactose Inhibits H-1PV Infection
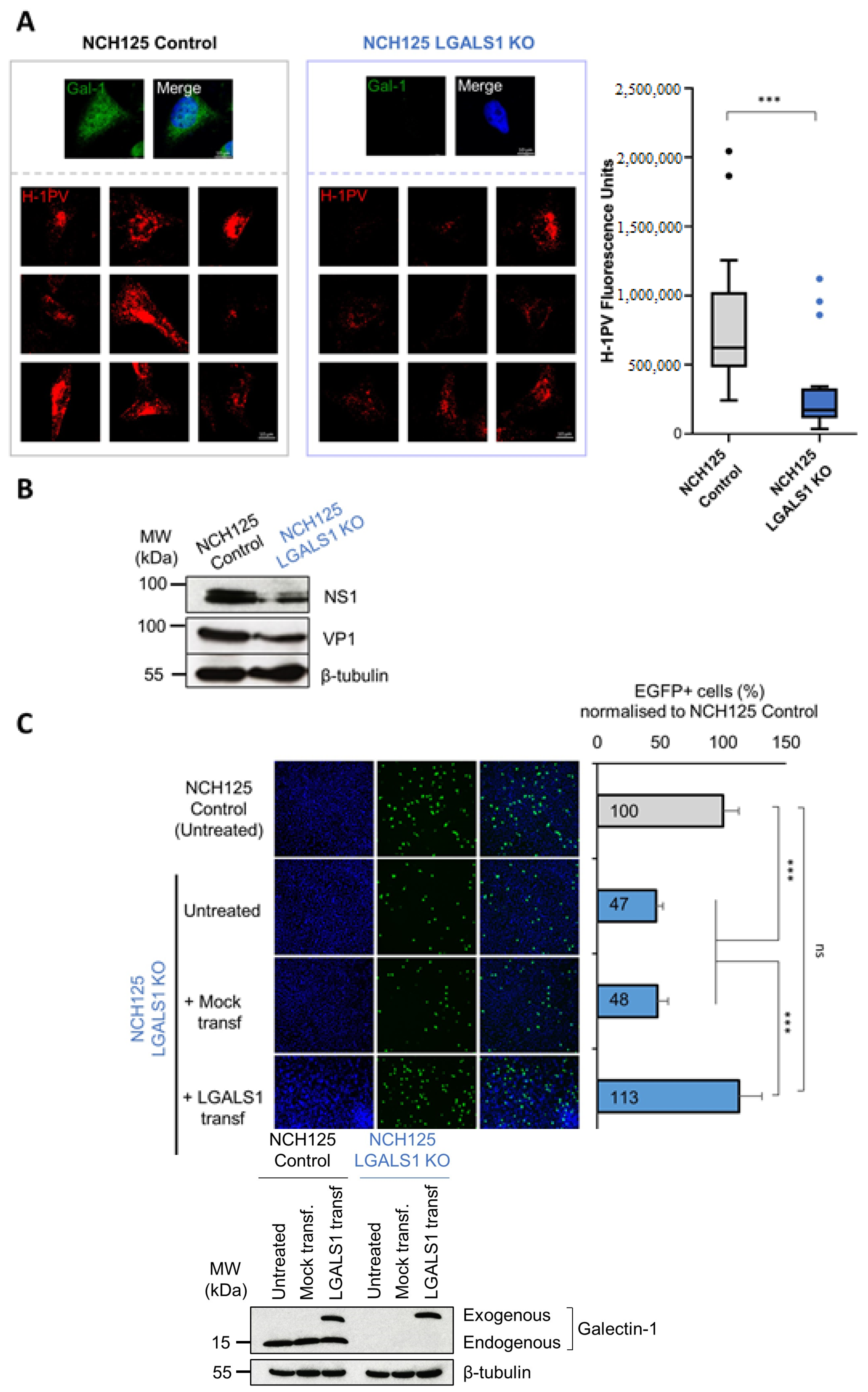
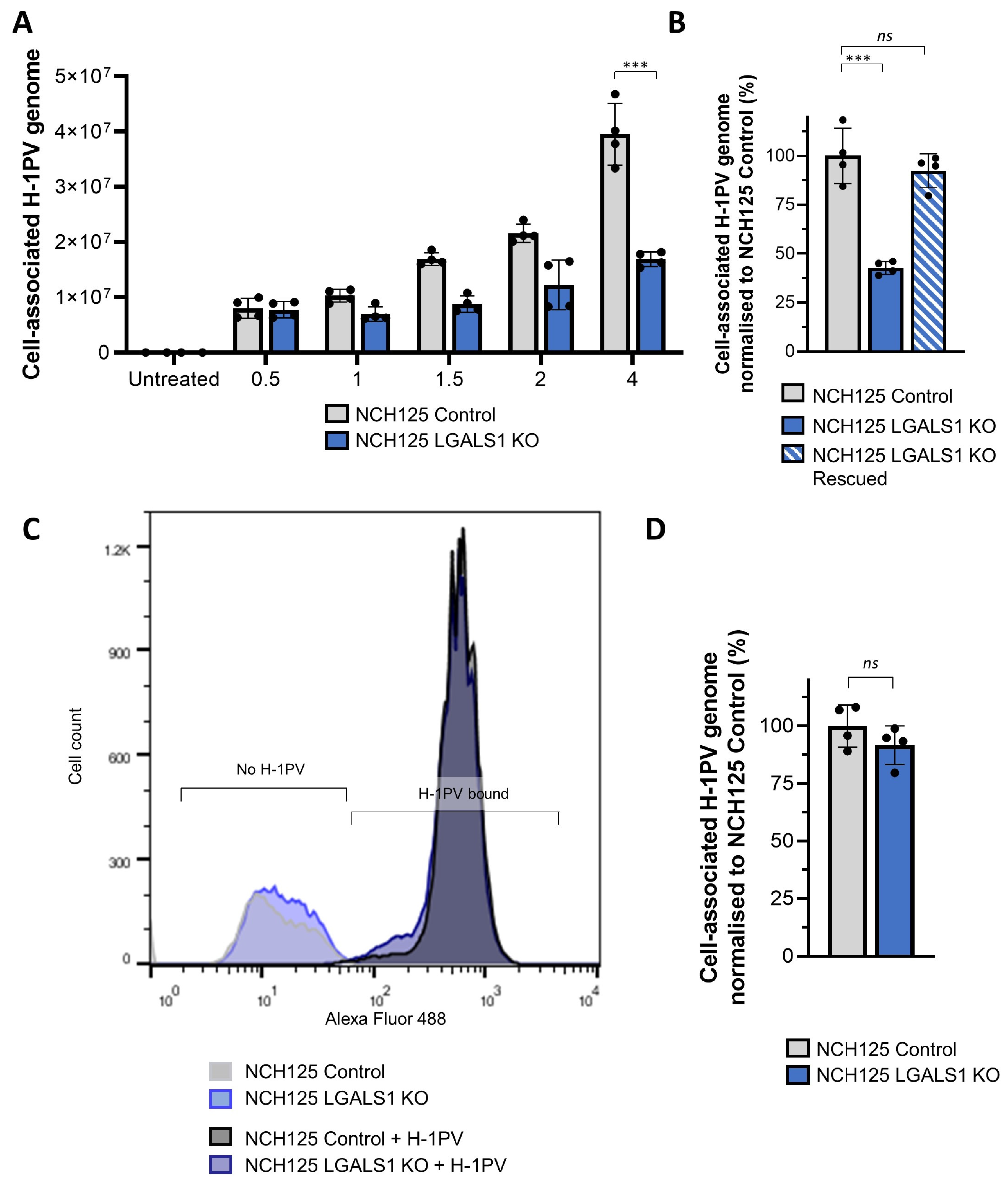
3.3. Galectin-1 Knockout Impairs H-1PV Infection in NCH125 Cells
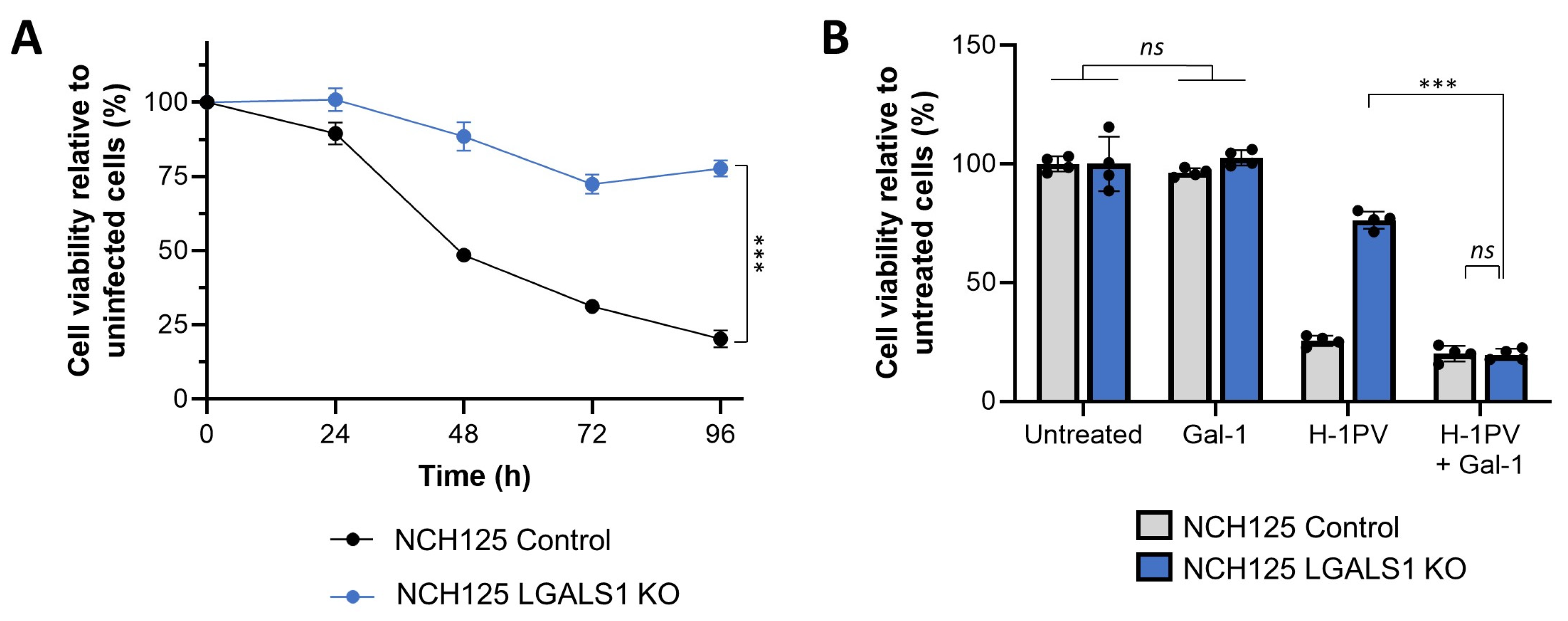
3.4. Galectin-1 Knockout Decreases H-1PV Oncolytic Activity in NCH125 Cells
3.5. Galectin-1 Plays a Role in H-1PV Virus Entry Rather Than Cell Surface Attachment
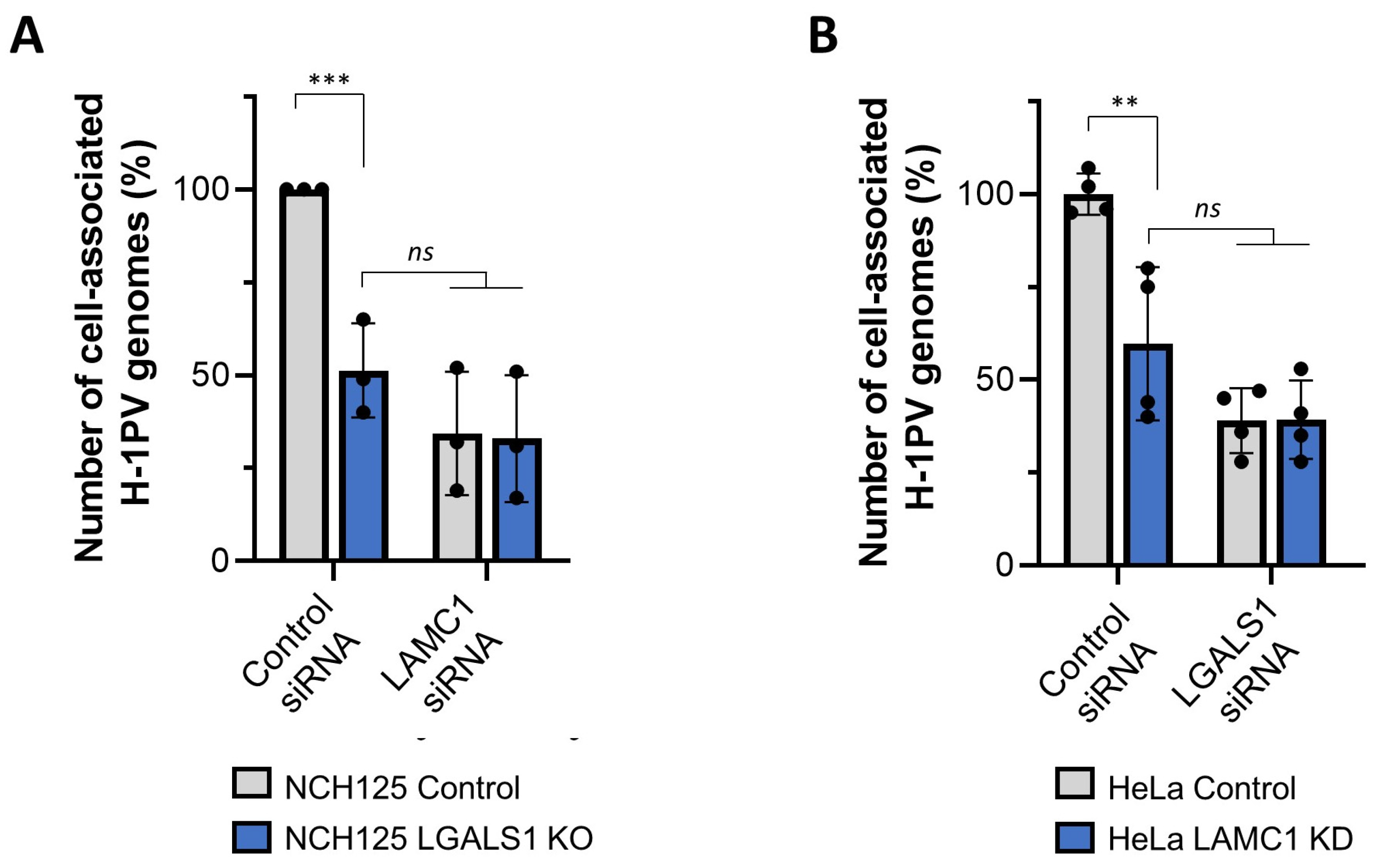
3.6. Gal-1 Cooperates with Laminins in Mediating H-1PV Infection
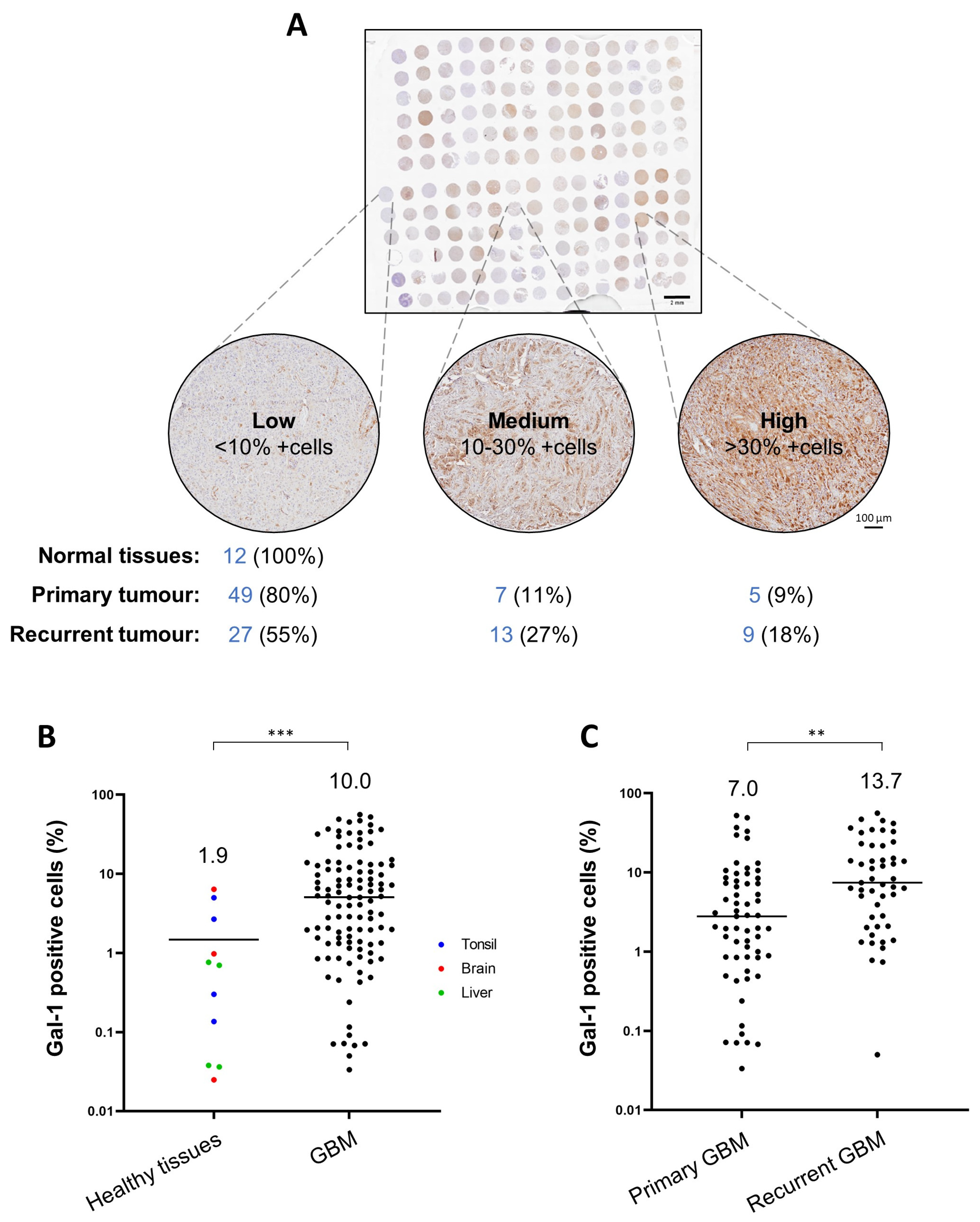
3.7. Gal-1 Is a Marker of Bad Prognosis in Various Tumour Types including GBM
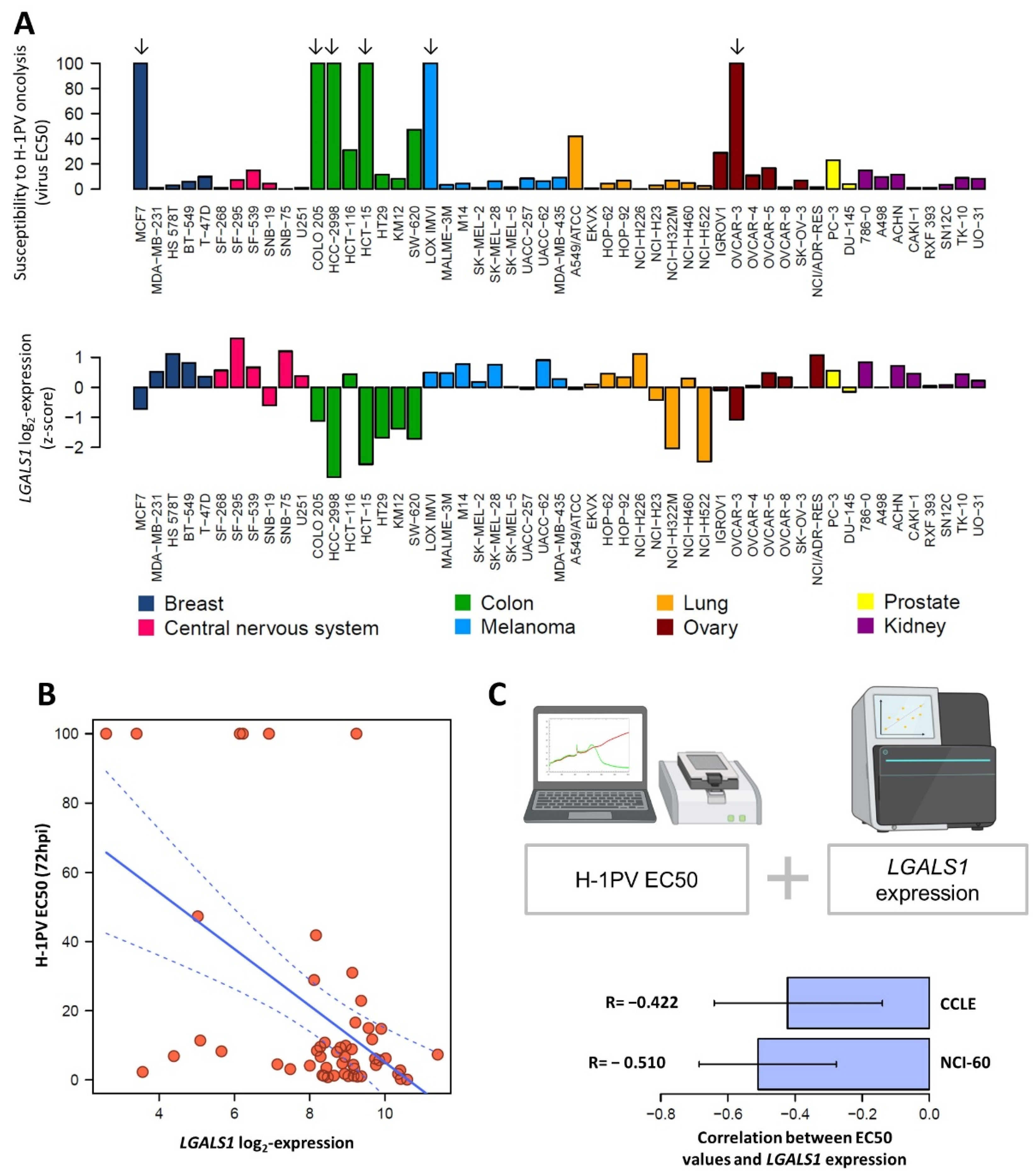
3.8. LGALS1 Expression Profile of NCI-60 Cells Positively Correlates with H-1PV Oncotoxicity
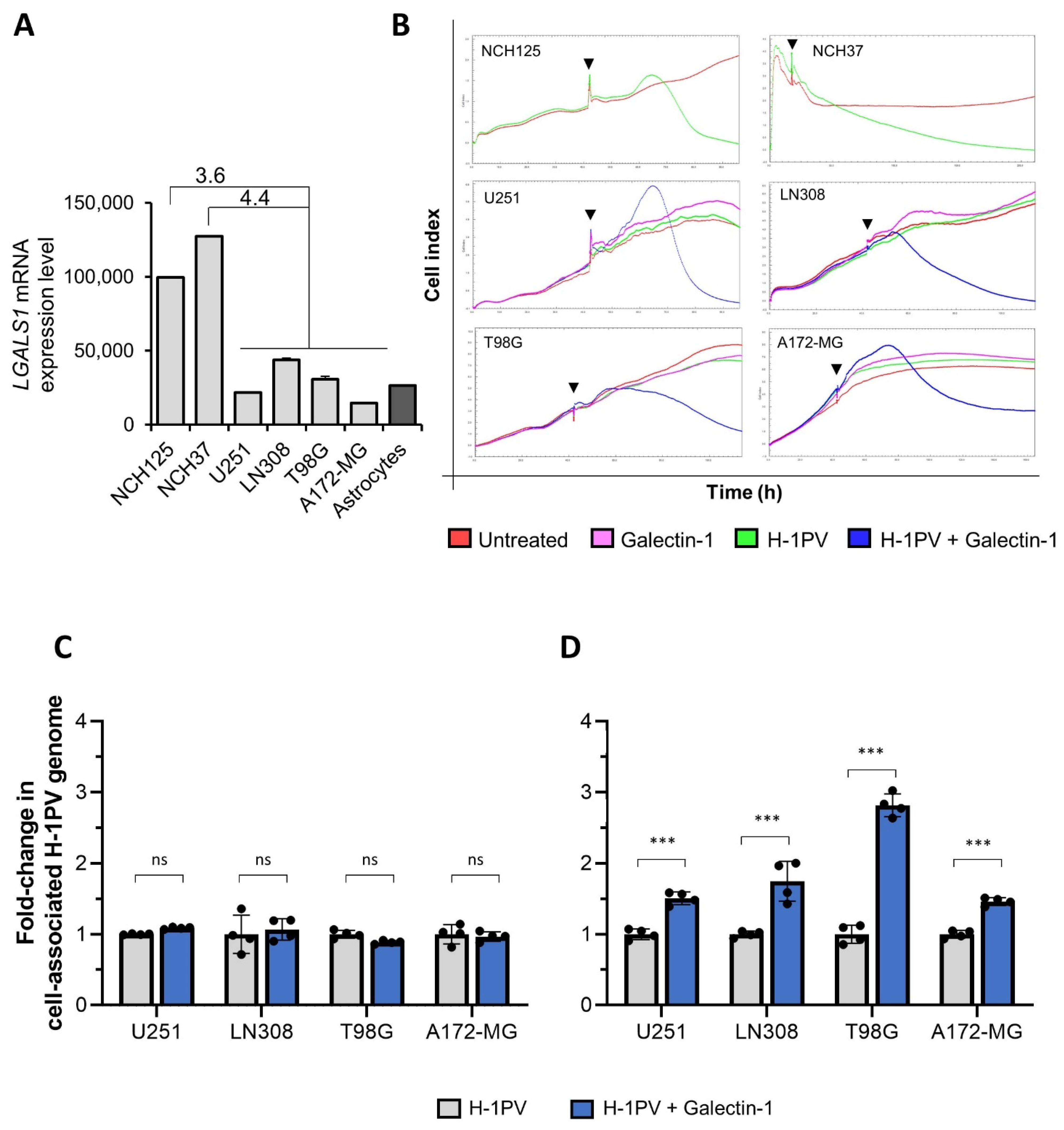
3.9. LGALS1 Expression Positively Correlates with H-1PV Oncolysis in Glioma Cell Lines
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchini, A.; Scott, E.M.; Rommelaere, J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses 2016, 8, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchini, A.; Daeffler, L.; Pozdeev, V.I.; Angelova, A.; Rommelaere, J. Immune Conversion of Tumor Microenvironment by Oncolytic Viruses: The Protoparvovirus H-1PV Case Study. Front. Immunol. 2019, 10, 1848. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007, 70, 183–232. [Google Scholar] [PubMed]
- Bretscher, C.; Marchini, A. H-1 parvovirus as a cancer-killing agent: Past, present, and future. Viruses 2019, 11, 562. [Google Scholar] [CrossRef] [Green Version]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D. The family parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef]
- Ros, C.; Bayat, N.; Wolfisberg, R.; Almendral, J.M.J.V. Protoparvovirus cell entry. Viruses 2017, 9, 313. [Google Scholar] [CrossRef]
- Hristov, G.; Kramer, M.; Li, J.; El-Andaloussi, N.; Mora, R.; Daeffler, L.; Zentgraf, H.; Rommelaere, J.; Marchini, A. Through Its Nonstructural Protein NS1, Parvovirus H-1 Induces Apoptosis via Accumulation of Reactive Oxygen Species. J. Virol. 2010, 84, 5909–5922. [Google Scholar] [CrossRef] [Green Version]
- Nuesch, J.P.; Lacroix, J.; Marchini, A.; Rommelaere, J. Molecular pathways: Rodent parvoviruses--mechanisms of oncolysis and prospects for clinical cancer treatment. Clin. Cancer Res. 2012, 18, 3516–3523. [Google Scholar] [CrossRef] [Green Version]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 12, 2620–2634. [Google Scholar] [CrossRef] [Green Version]
- Hajda, J.; Leuchs, B.; Angelova, A.L.; Frehtman, V.; Rommelaere, J.; Mertens, M.; Pilz, M.; Kieser, M.; Krebs, O.; Dahm, M.; et al. Phase 2 Trial of Oncolytic H-1 Parvovirus Therapy Shows Safety and Signs of Immune System Activation in Patients With Metastatic Pancreatic Ductal Adenocarcinoma. Clin. Cancer Res. 2021, 27, 5546–5556. [Google Scholar] [CrossRef]
- Hartley, A.; Kavishwar, G.; Salvato, I.; Marchini, A. A Roadmap for the Success of Oncolytic Parvovirus-Based Anticancer Therapies. Ann. Rev. Virol 2020, 7, 537–557. [Google Scholar] [CrossRef] [PubMed]
- Allaume, X.; El-Andaloussi, N.; Leuchs, B.; Bonifati, S.; Kulkarni, A.; Marttila, T.; Kaufmann, J.K.; Nettelbeck, D.M.; Kleinschmidt, J.; Rommelaere, J.; et al. Retargeting of rat parvovirus H-1PV to cancer cells through genetic engineering of the viral capsid. J. Virol. 2012, 86, 3452–3465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halder, S.; Nam, H.J.; Govindasamy, L.; Vogel, M.; Dinsart, C.; Salome, N.; McKenna, R.; Agbandje-McKenna, M. Structural characterization of H-1 parvovirus: Comparison of infectious virions to empty capsids. J. Virol. 2013, 87, 5128–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, A.; Ferreira, T.; Bretscher, C.; Grewenig, A.; El-Andaloussi, N.; Bonifati, S.; Marttila, T.; Palissot, V.; Hossain, J.A.; Azuaje, F.; et al. Oncolytic H-1 parvovirus binds to sialic acid on laminins for cell attachment and entry. Nat. Commun. 2021, 12, 3834. [Google Scholar] [CrossRef]
- Ferreira, T.; Kulkarni, A.; Bretscher, C.; Richter, K.; Ehrlich, M.; Marchini, A. Oncolytic H-1 Parvovirus Enters Cancer Cells through Clathrin-Mediated Endocytosis. Viruses 2020, 12, 1199. [Google Scholar] [CrossRef]
- Vandenbrule, F.; Buicu, C.; Baldet, M.; Sobel, M.E.; Cooper, D.N.; Marschal, P.; Castronovo, V. Galectin-1 modulates human melanoma cell adhesion to laminin. Biochem. Biophys. Res. Commun. 1995, 209, 760–767. [Google Scholar] [CrossRef]
- Cousin, J.M.; Cloninger, M.J. The role of galectin-1 in cancer progression, and synthetic multivalent systems for the study of galectin-1. Int. J. Mol. Sci. 2016, 17, 1566. [Google Scholar] [CrossRef] [Green Version]
- Vasta, G.R.; Ahmed, H.; Bianchet, M.A.; Fernández-Robledo, J.A.; Amzel, L.M. Diversity in recognition of glycans by F-type lectins and galectins: Molecular, structural, and biophysical aspects. Ann. N. Y. Acad. Sci. 2012, 1253, E14. [Google Scholar] [CrossRef] [Green Version]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci 2018, 131, jcs208884. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.H.; Lin, C.Y.; Chang, M.R.; Urbina, A.N.; Assavalapsakul, W.; Thitithanyanont, A.; Chen, Y.H.; Liu, F.T.; Wang, S.F. The role of galectins in virus infection—A systemic literature review. J. Microbiol. Immunol Infect. 2020, 53, 925–935. [Google Scholar] [CrossRef]
- Ouellet, M.; Mercier, S.; Pelletier, I.; Bounou, S.; Roy, J.; Hirabayashi, J.; Sato, S.; Tremblay, M.J. Galectin-1 acts as a soluble host factor that promotes HIV-1 infectivity through stabilization of virus attachment to host cells. J. Immunol. 2005, 174, 4120–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.-L.; Chen, Y.-H.; Wang, S.-W.; Huang, Y.-J.; Leu, C.-H.; Yeh, N.-C.; Chu, C.-Y.; Lin, C.-C.; Shieh, G.-S.; Chen, Y.-L. Galectin-1 binds to influenza virus and ameliorates influenza virus pathogenesis. J. Virol. 2011, 85, 10010–10020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcin, P.O.; Nabi, I.R.; Pante, N. Galectin-3 plays a role in minute virus of mice infection. Virology 2015, 481, 63–72. [Google Scholar] [CrossRef] [Green Version]
- Garcin, P.; Cohen, S.; Terpstra, S.; Kelly, I.; Foster, L.J.; Panté, N. Proteomic analysis identifies a novel function for galectin-3 in the cell entry of parvovirus. J. Proteom. 2013, 79, 123–132. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, N.; Endele, M.; Leuchs, B.; Bonifati, S.; Kleinschmidt, J.; Rommelaere, J.; Marchini, A. Novel adenovirus-based helper system to support production of recombinant parvovirus. Cancer Gene Ther. 2011, 18, 240–249. [Google Scholar] [CrossRef] [PubMed]
- El-Andaloussi, N.; Leuchs, B.; Bonifati, S.; Rommelaere, J.; Marchini, A. Efficient recombinant parvovirus production with the help of adenovirus-derived systems. J. Vis. Exp. 2012, 62, e3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodendorf, U.; Cziepluch, C.; Jauniaux, J.-C.; Rommelaere, J.; Salomé, N. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 1999, 73, 7769–7779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kestler, J.; Neeb, B.; Struyf, S.; Damme, J.V.; Cotmore, S.F.; D’Abramo, A.; Tattersall, P.; Rommelaere, J.; Dinsart, C.; Cornelis, J.J. cis requirements for the efficient production of recombinant DNA vectors based on autonomous parvoviruses. Hum. Gene Ther. 1999, 10, 1619–1632. [Google Scholar] [CrossRef]
- Leuchs, B.; Roscher, M.; Muller, M.; Kurschner, K.; Rommelaere, J. Standardized large-scale H-1PV production process with efficient quality and quantity monitoring. J. Virol. Methods 2016, 229, 48–59. [Google Scholar] [CrossRef] [Green Version]
- Hossain, J.A.; Riecken, K.; Miletic, H.; Fehse, B. Cancer suicide gene therapy with TK. 007. In Suicide Gene Therapy; Springer: Berlin/Heidelberg, Germany, 2019; pp. 11–26. [Google Scholar]
- Hossain, J.A.; Latif, M.A.; Ystaas, L.A.; Ninzima, S.; Riecken, K.; Muller, A.; Azuaje, F.; Joseph, J.V.; Talasila, K.M.; Ghimire, J. Long-term treatment with valganciclovir improves lentiviral suicide gene therapy of glioblastoma. J. Neurooncol. 2019, 21, 890–900. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Nesmelova, I.V.; Ermakova, E.; Daragan, V.A.; Pang, M.; Menendez, M.; Lagartera, L.; Solis, D.; Baum, L.G.; Mayo, K.H. Lactose binding to galectin-1 modulates structural dynamics, increases conformational entropy, and occurs with apparent negative cooperativity. J. Mol. Biol. 2010, 397, 1209–1230. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wu, T.; Wang, K.; Luo, S.; Chen, Z.; Fan, M.; Xue, D.; Lu, H.; Zhuang, Q.; Xu, X. Prognostic significance of galectin-1 expression in patients with cancer: A meta-analysis. Cancer Cell Int. 2018, 18, 108. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, R.H. The NCI60 human tumour cell line anticancer drug screen. Nat. Rev. Cancer 2006, 6, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Herrero y Calle, M.; Cornelis, J.J.; Herold-Mende, C.; Rommelaere, J.; Schlehofer, J.R.; Geletneky, K. Parvovirus H-1 infection of human glioma cells leads to complete viral replication and efficient cell killing. Int. J. Cancer 2004, 109, 76–84. [Google Scholar] [CrossRef]
- Angelova, A.; Ferreira, T.; Bretscher, C.; Rommelaere, J.; Marchini, A. Parvovirus-Based Combinatorial Immunotherapy: A Reinforced Therapeutic Strategy against Poor-Prognosis Solid Cancers. Cancers 2021, 13, 342. [Google Scholar] [CrossRef]
- Elola, M.T.; Wolfenstein-Todel, C.; Troncoso, M.F.; Vasta, G.R.; Rabinovich, G.A. Galectins: Matricellular glycan-binding proteins linking cell adhesion, migration, and survival. Cell Mol. Life Sci. 2007, 64, 1679–1700. [Google Scholar] [CrossRef]
- Bänfer, S.; Jacob, R. Galectins in Intra-and Extracellular Vesicles. Biomolecules 2020, 10, 1232. [Google Scholar] [CrossRef]
- Fajka-Boja, R.; Blasko, A.; Kovacs-Solyom, F.; Szebeni, G.; Toth, G.; Monostori, E. Co-localization of galectin-1 with GM1 ganglioside in the course of its clathrin-and raft-dependent endocytosis. Cell Mol. Life Sci. 2008, 65, 2586–2593. [Google Scholar] [CrossRef]
- Lepur, A.; Carlsson, M.C.; Novak, R.; Dumić, J.; Nilsson, U.J.; Leffler, H. Galectin-3 endocytosis by carbohydrate independent and dependent pathways in different macrophage like cell types. Biochim. Biophy. Acta Gen. Subj. 2012, 1820, 804–818. [Google Scholar] [CrossRef]
- Brewer, C.F. Binding and cross-linking properties of galectins. Biochim. Biophys. Acta Gen. Subj. 2002, 1572, 255–262. [Google Scholar] [CrossRef]
- Garner, O.B.; Baum, L.G. Galectin–glycan lattices regulate cell-surface glycoprotein organization and signalling. Bioch. Soc. Trans. 2008, 36, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- Pace, K.E.; Lee, C.; Stewart, P.L.; Baum, L.G. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J. Immunol. 1999, 163, 3801–3811. [Google Scholar]
- Moiseeva, E.P.; Williams, B.; Samani, N.J. Galectin 1 inhibits incorporation of vitronectin and chondroitin sulfate B into the extracellular matrix of human vascular smooth muscle cells. Biochim. Biophys. Acta Gen. Subj. 2003, 1619, 125–132. [Google Scholar] [CrossRef]
- Moiseeva, E.P.; Javed, Q.; Spring, E.L.; de Bono, D.P. Galectin 1 is involved in vascular smooth muscle cell proliferation. Cardiovasc. Res. 2000, 45, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Mercier, S.; St-Pierre, C.; Pelletier, I.; Ouellet, M.; Tremblay, M.J.; Sato, S. Galectin-1 promotes HIV-1 infectivity in macrophages through stabilization of viral adsorption. Virology 2008, 371, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Lee, P.-H.; Liu, C.-M.; Ho, T.-S.; Tsai, Y.-C.; Lin, C.-C.; Wang, Y.-F.; Chen, Y.-L.; Yu, C.-K.; Wang, S.-M.; Liu, C.-C. Enterovirus 71 virion-associated galectin-1 facilitates viral replication and stability. PLoS ONE 2015, 10, e0116278. [Google Scholar] [CrossRef] [Green Version]
- Garner, O.B.; Yun, T.; Pernet, O.; Aguilar, H.C.; Park, A.; Bowden, T.A.; Freiberg, A.N.; Lee, B.; Baum, L.G. Timing of galectin-1 exposure differentially modulates Nipah virus entry and syncytium formation in endothelial cells. J. Virol. 2015, 89, 2520–2529. [Google Scholar] [CrossRef] [Green Version]
- Levroney, E.L.; Aguilar, H.C.; Fulcher, J.A.; Kohatsu, L.; Pace, K.E.; Pang, M.; Gurney, K.B.; Baum, L.G.; Lee, B. Novel innate immune functions for galectin-1: Galectin-1 inhibits cell fusion by Nipah virus envelope glycoproteins and augments dendritic cell secretion of proinflammatory cytokines. J. Immunol. 2005, 175, 413–420. [Google Scholar] [CrossRef] [Green Version]
- Garner, O.B.; Aguilar, H.C.; Fulcher, J.A.; Levroney, E.L.; Harrison, R.; Wright, L.; Robinson, L.R.; Aspericueta, V.; Panico, M.; Haslam, S.M. Endothelial galectin-1 binds to specific glycans on nipah virus fusion protein and inhibits maturation, mobility, and function to block syncytia formation. PLoS Pathog. 2010, 6, e1000993. [Google Scholar] [CrossRef]
- Chou, F.-C.; Chen, H.-Y.; Kuo, C.-C.; Sytwu, H.-K.J. Role of galectins in tumors and in clinical immunotherapy. Int. J. Mol. Sci. 2018, 19, 430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Zhou, S.-J.; Zhang, Y.; Zhang, G.-Q.; Zha, T.-Z.; Feng, Y.-Z.; Zhang, K. Clinicopathological and prognostic significance of galectin-1 and vascular endothelial growth factor expression in gastric cancer. World J. Gastroenterol. 2013, 19, 2073. [Google Scholar] [CrossRef] [PubMed]
- Schulz, H.; Schmoeckel, E.; Kuhn, C.; Hofmann, S.; Mayr, D.; Mahner, S.; Jeschke, U. Galectins-1,-3, and-7 are prognostic markers for survival of ovarian cancer patients. Int. J. Mol. Sci. 2017, 18, 1230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Bosch, N.; Barranco, L.E.; Orozco, C.A.; Moreno, M.; Visa, L.; Iglesias, M.; Oldfield, L.; Neoptolemos, J.P.; Greenhalf, W.; Earl, J. Increased plasma levels of galectin-1 in pancreatic cancer: Potential use as biomarker. Oncotarget 2018, 9, 32984. [Google Scholar] [CrossRef] [Green Version]
- Chou, S.-Y.; Yen, S.-L.; Huang, C.-C.; Huang, E.-Y. Galectin-1 is a poor prognostic factor in patients with glioblastoma multiforme after radiotherapy. BMC Cancer 2018, 18, 105. [Google Scholar] [CrossRef] [Green Version]
- Jung, T.-Y.; Jung, S.; Ryu, H.-H.; Jeong, Y.-I.; Jin, Y.-H.; Jin, S.-G.; Kim, I.-Y.; Kang, S.-S.; Kim, H.-S. Role of galectin-1 in migration and invasion of human glioblastoma multiforme cell lines. J. Neurosurg. 2008, 109, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Verschuere, T.; Van Woensel, M.; Fieuws, S.; Lefranc, F.; Mathieu, V.; Kiss, R.; Van Gool, S.W.; De Vleeschouwer, S. Altered galectin-1 serum levels in patients diagnosed with high-grade glioma. J. Neurooncol. 2013, 115, 9–17. [Google Scholar] [CrossRef]
- Astorgues-Xerri, L.; Riveiro, M.E.; Tijeras-Raballand, A.; Serova, M.; Neuzillet, C.; Albert, S.; Raymond, E.; Faivre, S. Unraveling galectin-1 as a novel therapeutic target for cancer. Cancer Treat. Rev. 2014, 40, 307–319. [Google Scholar] [CrossRef]
- Blanchard, H.; Bum-Erdene, K.; Bohari, M.H.; Yu, X. Galectin-1 inhibitors and their potential therapeutic applications: A patent review. Expert Opin. Ther. Pat. 2016, 26, 537–554. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, T.; Kulkarni, A.; Bretscher, C.; Nazarov, P.V.; Hossain, J.A.; Ystaas, L.A.R.; Miletic, H.; Röth, R.; Niesler, B.; Marchini, A. Oncolytic H-1 Parvovirus Hijacks Galectin-1 to Enter Cancer Cells. Viruses 2022, 14, 1018. https://doi.org/10.3390/v14051018
Ferreira T, Kulkarni A, Bretscher C, Nazarov PV, Hossain JA, Ystaas LAR, Miletic H, Röth R, Niesler B, Marchini A. Oncolytic H-1 Parvovirus Hijacks Galectin-1 to Enter Cancer Cells. Viruses. 2022; 14(5):1018. https://doi.org/10.3390/v14051018
Chicago/Turabian StyleFerreira, Tiago, Amit Kulkarni, Clemens Bretscher, Petr V. Nazarov, Jubayer A. Hossain, Lars A. R. Ystaas, Hrvoje Miletic, Ralph Röth, Beate Niesler, and Antonio Marchini. 2022. "Oncolytic H-1 Parvovirus Hijacks Galectin-1 to Enter Cancer Cells" Viruses 14, no. 5: 1018. https://doi.org/10.3390/v14051018