
Insights into Gene Transcriptional Regulation of Kayvirus Bacteriophages Obtained from Therapeutic Mixtures


 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Results
2.1. Host Range of Staphylococcus Phages SAM1 and SAM2
2.2. Genomic Features of Phages SAM1 and SAM2
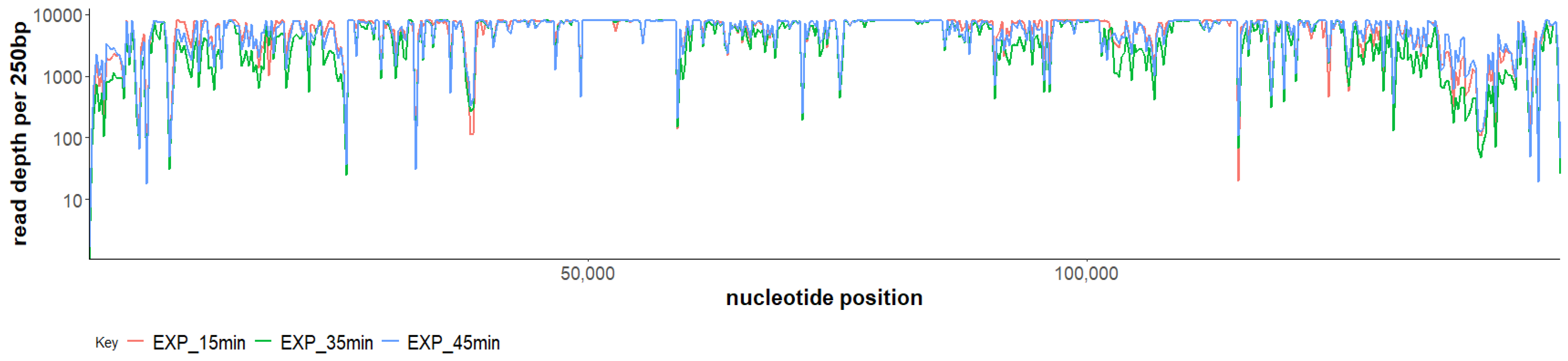
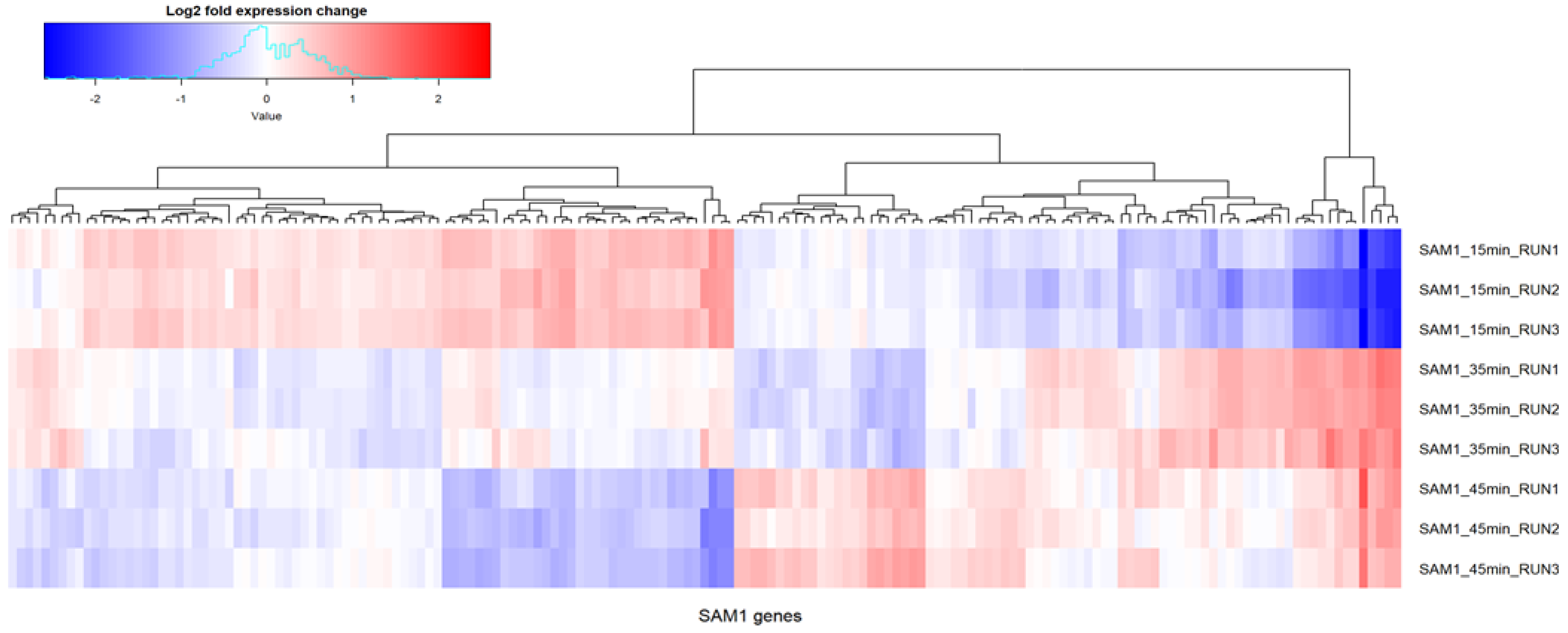
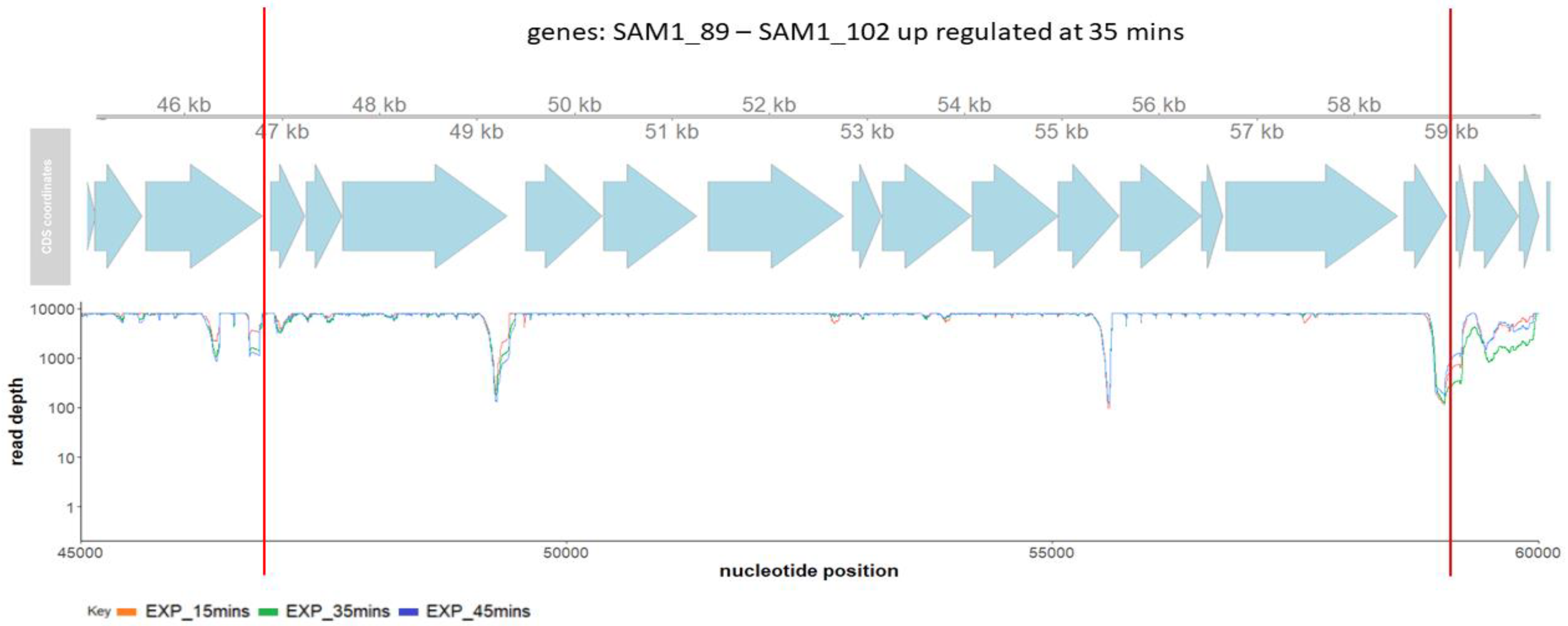
2.3. Phage SAM1 Gene Transcription Analysis
2.4. Host Gene Transcription Analysis during Phage SAM1 Infection
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Phage, and Growth Requirements
4.2. Phage Host Range
4.3. DNA Extraction, Sequencing, and Genome Assembly
4.4. Genome Annotation and Comparison
4.5. RNA-seq Analysis of Phage SAM2
4.6. Accession Numbers for Sequence Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. The complete genomes and proteomes of 27 Staphylococcus aureus bacteriophages. Proc. Natl. Acad. Sci. USA 2005, 102, 5174–5179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.S.; De Lencastre, H.; Garau, J.; Kluytmans, J.; Malhotra-Kumar, S.; Peschel, A.; Harbarth, S. Methicillin-resistant Staphylococcus aureus. Nat. Rev. Dis. Prim. 2018, 4, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Xia, G.; Wolz, C. Phages of Staphylococcus aureus and their impact on host evolution. Infect. Genet. Evol. 2014, 21, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [Green Version]
- Łobocka, M.; Hejnowicz, M.S.; Dąbrowski, K.; Gozdek, A.; Kosakowski, J.; Witkowska, M.; Ulatowska, M.I.; Weber-Dąbrowska, B.; Kwiatek, M.; Parasion, S.; et al. Genomics of staphylococcal Twort-like phages--potential therapeutics of the post-antibiotic era. Adv. Virus Res. 2012, 83, 143–216. [Google Scholar] [PubMed]
- Botka, T.; Pantůček, R.; Mašlaňová, I.; Benešík, M.; Petráš, P.; Růžičková, V.; Havlíčková, P.; Varga, M.; Žemličková, H.; Koláčková, I.; et al. Lytic and genomic properties of spontaneous host-range Kayvirus mutants prove their suitability for upgrading phage therapeutics against staphylococci. Sci. Rep. 2019, 9, 5475. [Google Scholar] [CrossRef]
- Melo, L.D.R.; Brandão, A.; Akturk, E.; Santos, S.B.; Azeredo, J. Characterization of a new Staphylococcus aureus Kayvirus harboring a lysin active against biofilms. Viruses 2018, 10, 182. [Google Scholar] [CrossRef] [Green Version]
- Abdelkader, K.; Gerstmans, H.; Saafan, A.; Dishisha, T.; Briers, Y. The preclinical and clinical progress of bacteriophages and their lytic enzymes: The parts are easier than the whole. Viruses 2019, 11, 96. [Google Scholar] [CrossRef] [Green Version]
- El Haddad, L.; Ben Abdallah, N.; Plante, P.-L.; Dumaresq, J.; Katsarava, R.; Labrie, S.; Corbeil, J.; St-Gelais, D.; Moineau, S. Improving the Safety of Staphylococcus aureus Polyvalent Phages by Their Production on a Staphylococcus xylosus Strain. PLoS ONE 2014, 9, e102600. [Google Scholar] [CrossRef] [Green Version]
- McCallin, S.; Sarker, S.A.; Sultana, S.; Oechslin, F.; Brüssow, H. Metagenome analysis of Russian and Georgian Pyophage cocktails and a placebo-controlled safety trial of single phage versus phage cocktail in healthy Staphylococcus aureus carriers. Environ. Microbiol. 2018, 20, 3278–3293. [Google Scholar] [CrossRef]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent Diabetic toe ulcers: A case series. J. Wound Care 2016, 25, S27–S33. [Google Scholar] [CrossRef]
- Onsea, J.; Soentjens, P.; Djebara, S.; Merabishvili, M.; Depypere, M.; Spriet, I.; De Munter, P.; Debaveye, Y.; Nijs, S.; Vanderschot, P.; et al. Bacteriophage Application for Difficult-To-Treat Musculoskeletal Infections: Development of a Standardized Multidisciplinary Treatment Protocol. Viruses 2019, 11, 891. [Google Scholar] [CrossRef] [Green Version]
- Adriaenssens, E.M.; Brister, J.R. How to Name and Classify Your Phage: An Informal Guide. Viruses 2017, 9, 70. [Google Scholar] [CrossRef] [Green Version]
- Ajuebor, J.; Buttimer, C.; Arroyo-Moreno, S.; Chanishvili, N.; Gabriel, E.M.; O’Mahony, J.; McAuliffe, O.; Neve, H.; Franz, C.; Coffey, A.; et al. Comparison of Staphylococcus phage K with close phage relatives commonly employed in phage therapeutics. Antibiotics 2018, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornienko, M.; Fisunov, G.; Bespiatykh, D.; Kuptsov, N.; Gorodnichev, R.; Klimina, K.; Kulikov, E.; Ilina, E.; Letarov, A.; Shitikov, E. Transcriptional landscape of Staphylococcus aureus Kayvirus Bacteriophage vB_SauM-515A1. Viruses 2020, 12, 1320. [Google Scholar] [CrossRef]
- Xia, G.; Corrigan, R.M.; Winstel, V.; Goerke, C.; Gründling, A.; Peschel, A. Wall teichoic acid-dependent adsorption of staphylococcal siphovirus and myovirus. J. Bacteriol. 2011, 193, 4006–4009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canchaya, C.; Fournous, G.; Chibani-Chennoufi, S.; Dillmann, M.L.; Brüssow, H. Phage as agents of lateral gene transfer. Curr. Opin. Microbiol. 2003, 6, 417–424. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Witney, A.A.; Lindsay, J.A. Staphylococcus aureus temperate bacteriophage: Carriage and horizontal gene transfer is lineage associated. Front. Cell. Infect. Microbiol. 2012, 2, 6. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Ram, G.; Penadés, J.R.; Brown, S.; Novick, R.P. Pathogenicity island-directed transfer of unlinked chromosomal virulence genes. Mol. Cell 2015, 57, 138–149. [Google Scholar] [CrossRef] [Green Version]
- Kaya, H.; Hasman, H.; Larsen, J.; Stegger, M.; Johannesen, T.B.; Allesøe, R.L.; Lemvigh, C.K.; Aarestrup, F.M.; Lund, O.; Larsen, A.R. SCC mec Finder, a Web-Based Tool for Typing of Staphylococcal Cassette Chromosome mec in Staphylococcus aureus Using Whole-Genome Sequence Data. Msphere 2018, 3, e00612-17. [Google Scholar] [CrossRef] [Green Version]
- Enright, M.C.; Day, N.P.J.; Davies, C.E.; Peacock, S.J.; Spratt, B.G. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. J. Clin. Microbiol. 2000, 38, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Rajewska, M.; Wegrzyn, K.; Konieczny, I. AT-rich region and repeated sequences—The essential elements of replication origins of bacterial replicons. FEMS Microbiol. Rev. 2012, 36, 408–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, D.; Schuch, R.; Chahales, P.; Zhu, S.; Fischetti, V.A. PlyC: A multimeric bacteriophage lysin. Proc. Natl. Acad. Sci. USA 2006, 103, 10765–10770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasdel, B.G.; Chevallereau, A.; Monot, M.; Lavigne, R.; Debarbieux, L. Comparative transcriptomics analyses reveal the conservation of an ancestral infectious strategy in two bacteriophage genera. ISME J. 2017, 11, 1988–1996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevallereau, A.; Blasdel, B.G.; De Smet, J.; Monot, M.; Zimmermann, M.; Kogadeeva, M.; Sauer, U.; Jorth, P.; Whiteley, M.; Debarbieux, L.; et al. Next-Generation “-omics” Approaches Reveal a Massive Alteration of Host RNA Metabolism during Bacteriophage Infection of Pseudomonas aeruginosa. PLoS Genet. 2016, 12, e1006134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, C.R.; Casjens, S.R.; Cresawn, S.G.; Houtz, J.M.; Smith, A.L.; Ford, M.E.; Peebles, C.L.; Hatfull, G.F.; Hendrix, R.W.; Huang, W.M.; et al. The genome of Bacillus subtilis bacteriophage SPO1. J. Mol. Biol. 2009, 388, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondy-Denomy, J.; Davidson, A.R. When a Virus is not a Parasite: The Beneficial Effects of Prophages on Bacterial Fitness. J. Microbiol. 2014, 52, 235–242. [Google Scholar] [CrossRef]
- Goerke, C.; Koller, J.; Wolz, C. Ciprofloxacin and Trimethoprim Cause Phage Induction and Virulence Modulation in Staphylococcus aureus. Antimicrob. Agents Chemother. 2006, 50, 171. [Google Scholar] [CrossRef] [Green Version]
- Petrovic Fabijan, A.; Lin, R.C.Y.; Ho, J.; Maddocks, S.; Ben Zakour, N.L.; Iredell, J.R.; Khalid, A.; Venturini, C.; Chard, R.; Morales, S.; et al. Safety of bacteriophage therapy in severe Staphylococcus aureus infection. Nat. Microbiol. 2020, 5, 465–472. [Google Scholar] [CrossRef]
- O’Flaherty, S.; Ross, R.P.; Meaney, W.; Fitzgerald, G.F.; Elbreki, M.F.; Coffey, A. Potential of the polyvalent anti-Staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl. Environ. Microbiol. 2005, 71, 1836–1842. [Google Scholar] [CrossRef] [Green Version]
- O’Flaherty, S.; Coffey, A.; Edwards, R.; Meaney, W.; Fitzgerald, G.F.; Ross, R.P. Genome of staphylococcal phage K: A new lineage of Myoviridae infecting gram-positive bacteria with a low G+C content. J. Bacteriol. 2004, 186, 2862–2871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonilla, N.; Rojas, M.I.; Cruz, G.N.F.; Hung, S.H.; Rohwer, F.; Barr, J.J. Phage on tap-a quick and efficient protocol for the preparation of bacteriophage laboratory stocks. PeerJ 2016, 4, e2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, D.; Vandenheuvel, D.; Martínez, B.; Rodríguez, A.; Lavigne, R.; García, P. Two Phages, phiIPLA-RODI and phiIPLA-C1C, Lyse Mono- and Dual-Species Staphylococcal Biofilms. Appl. Environ. Microbiol. 2015, 81, 3336–3348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.M.; Lohse, M.; Usadel, B. Genome analysis Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.A.; Dvorkin, M.; Kulikov, A.S.S.; Lesin, V.M.M.; Nikolenko, S.I.I.; Pham, S.; Prjibelski, A.D.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Söding, J.; Biegert, A.; Lupas, A.N.; Soding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juncker, A.S.; Willenbrock, H.; von Heijne, G.; Brunak, S.; Nielsen, H.; Krogh, A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003, 12, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Käll, L.; Krogh, A.; Sonnhammer, E.L.L. A combined transmembrane topology and signal peptide prediction method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Vandersteegen, K.; Mattheus, W.; Ceyssens, P.J.; Bilocq, F.; de Vos, D.; Pirnay, J.P.; Noben, J.P.; Merabishvili, M.; Lipinska, U.; Hermans, K.; et al. Microbiological and Molecular Assessment of Bacteriophage ISP for the Control of Staphylococcus aureus. PLoS ONE 2011, 6, e24418. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.; Findeiß, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (Co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; Dicuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| S. aureus Strain | SCCmec Type | MLST | Bacteriophages | |
|---|---|---|---|---|
| SAM1 | SAM2 | |||
| EOP ± SD | EOP ± SD | |||
| DPC5246 | Not detected | 71 | 1.2 ± 0.4 | 3.72 ± 0.48 |
| 0.0066(IIv)ST239 | II(2A) ** | 36 | 5.1 × 10−1 ± 0.1 | 4.37 ± 1.8 |
| 0.1206(IV)ST250 | IVc(2B) | 12 | 5.74 × 10−1 ± 0.4 | no plaques |
| 0.1239(III)ST239 | III(3A) | 239 | 1.4 × 10−1 ± 0.05 | 6.6 ± 2.46 |
| 0.1345(II)ST5 | VIII(4A) | 8 | 2.23 × 10−1 ±0.1 | 1.96 ± 0.51 |
| 0073(III)ST239 | III(3A) | 239 | 1.21 × 10−1 ± 0.1 | no plaques |
| 0104(III)ST239 | III(3A) | 239 | 9.65 × 10−2 ± 0.01 | 3.5 ± 1.19 |
| 0220(II)ST5 | IV(2B) ** | 8 | 2.22 × 10−2 ± 0.003 | 14.68 ± 4.31 |
| 0242(IV)ST30 | II(2A) | 496 | 1.95 × 10−1 ± 0.17 | 9.35 ± 3.15 |
| 0308(IA)ST247 | I(1B) | 247 | 3.41 × 10−1 ± 0.26 | 11.85 ± 3.24 |
| 3045(IIv)ST8 | II(2A) | 8 | 1.94 × 10−1 ± 0.14 | 4.54 ± 2.13 |
| 3144(IIv)ST8 | II(2A) | 8 | 2,17 × 10−1 ± 0.22 | 10.46 ± 5.18 |
| 3488(vv)ST8 | IV(2B) ** | 8 | 6.72 × 10−2 ± 0.01 | 11.11 ± 4.35 |
| 3581(IA)ST247 | VIII(4A) ** | 8 | 1 × 10−1 ± 0.01 | 5.51 ± 1.25 |
| 3594(II)ST36 | III(3A) | 239 | 5.77 × 10−1 ± 0.5 | 75.47 ± 2.32 |
| 3596(IIv)ST8 | VIII(4A) ** | 8 | 2.29 × 10−1 ± 0.05 | 28.89 ± 16.48 |
| E1038(IIv)ST8 | II(2A) | 8 | 9.7 × 10−1 ± 0.97 | 13.47 ± 3.47 |
| E1139(IV)ST45 | IVa(2B) | 45 | no plaques | 1 * |
| E1174(IV)ST22 | IV(2B) | 22 | 1.4 × 10−1 ± 0.01 | 7.37 × 10−1 |
| E1185(IV)ST12 | IVc | 12 | 1 * | 3.5 × 10−4 ± 3.3 × 10−4 |
| E1202(II)ST496 | VIII(4A) ** | 8 | 3.59 × 10−1 ± 0.2 | 20.83 ± 3.24 |
| M03/0073(III)ST239 | III(3A) | 239 | 1.32 ± 0.7 | 16.54 ± 0.52 |
| 0104(III)ST239 | III(3A) | 239 | 1.2 ± 0.4 | 3.72 ± 0.48 |
| 0220(II)ST5 | IV(2B) ** | 8 | 5.1 × 10−1 ± 0.1 | 4.37 ± 1.8 |
| 0242(IV)ST30 | II(2A) | 496 | 5.74 × 10−1 ± 0.4 | no plaques |
| Virulence Factors (Locus_tag, Product, Gene) | log2 Fold Change | ||
|---|---|---|---|
| 15 min | 35 min | 45 min | |
| LUU82_11680—bi-component gamma-hemolysin HlgCB subunit C—hlgC | −0.11173 | −0.0293 | 0.141026 |
| LUU82_04645—alpha-hemolysin—hyl | −0.15039 | −0.04266 | 0.193054 |
| LUU82_09700—staphylococcal protein A—spa | −0.26288 | −0.0223 | 0.285182 |
| LUU82_09135—complement inhibitor SCIN—scn | −0.18095 | −0.16701 | 0.347955 |
| LUU82_04665—complement convertase inhibitor—efb | −0.44123 | −0.06633 | −0.507566 |
| LUU82_00160—extracellular adherence protein Eap/Map—eap | −0.18624 | −0.03037 | 0.216609 |
| LUU82_11260—fibronectin-binding protein FnbA—fnbA | −0.43413 | −0.09529 | 0.529417 |
| LUU82_02360—staphylococcal enterotoxin type—sel26 | 0.415231 | −0.08836 | −0.32687 |
| LUU82_09885—staphylococcal enterotoxin type Z—selZ | 0.413472 | −0.08253 | −0.33094 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo-Moreno, S.; Buttimer, C.; Bottacini, F.; Chanishvili, N.; Ross, P.; Hill, C.; Coffey, A. Insights into Gene Transcriptional Regulation of Kayvirus Bacteriophages Obtained from Therapeutic Mixtures. Viruses 2022, 14, 626. https://doi.org/10.3390/v14030626
Arroyo-Moreno S, Buttimer C, Bottacini F, Chanishvili N, Ross P, Hill C, Coffey A. Insights into Gene Transcriptional Regulation of Kayvirus Bacteriophages Obtained from Therapeutic Mixtures. Viruses. 2022; 14(3):626. https://doi.org/10.3390/v14030626
Chicago/Turabian StyleArroyo-Moreno, Sara, Colin Buttimer, Francesca Bottacini, Nina Chanishvili, Paul Ross, Colin Hill, and Aidan Coffey. 2022. "Insights into Gene Transcriptional Regulation of Kayvirus Bacteriophages Obtained from Therapeutic Mixtures" Viruses 14, no. 3: 626. https://doi.org/10.3390/v14030626