
Regulation of Gene Expression of phiEco32-like Bacteriophage 7-11


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Comparison of phiEco32-like Genomes and Prediction of Their Promoters
2.2. Bacterial Strains, Phage and Growth Conditions
2.3. RNA Purification
2.4. Primer Extension Analysis
2.5. 7-11 Gene 47 Cloning and SaPh711_gp47 Purification
2.6. In Vitro Transcription
2.7. KmnO4 Probing
2.8. Fluorometric Measurements
3. Results
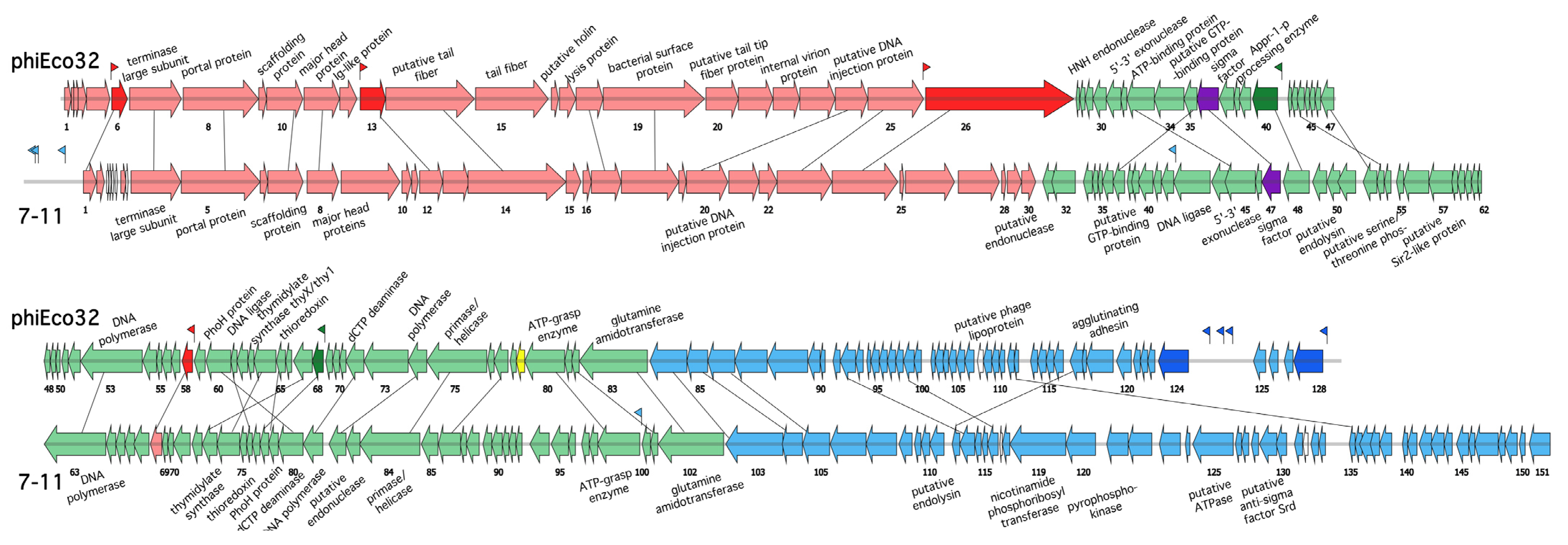
3.1. The phiEco32-like Phages and Their Genomes
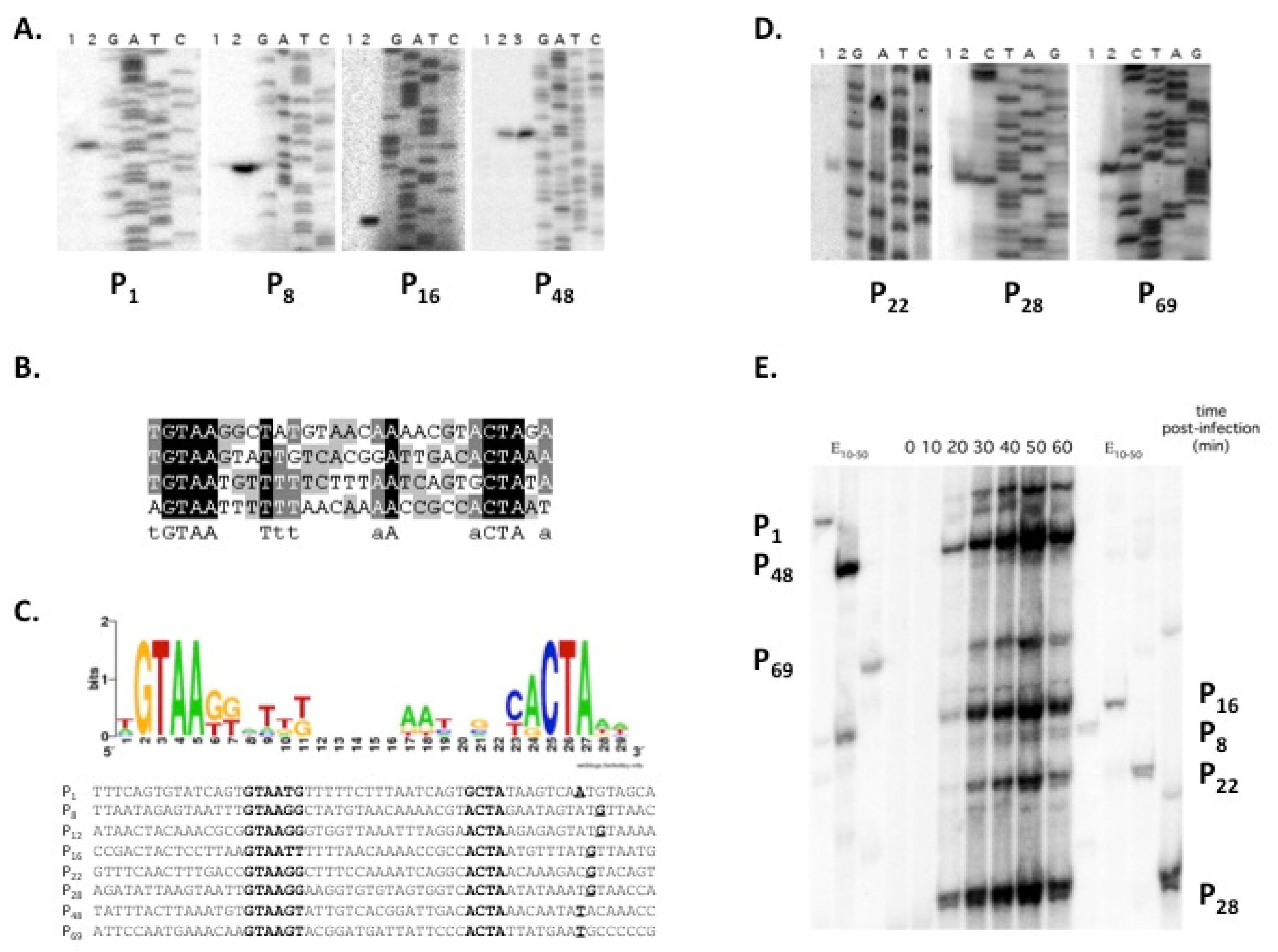
3.2. Identification of 7-11 Late Promoters
3.3. SaPh711_gp47 as a Late Sigma Factor
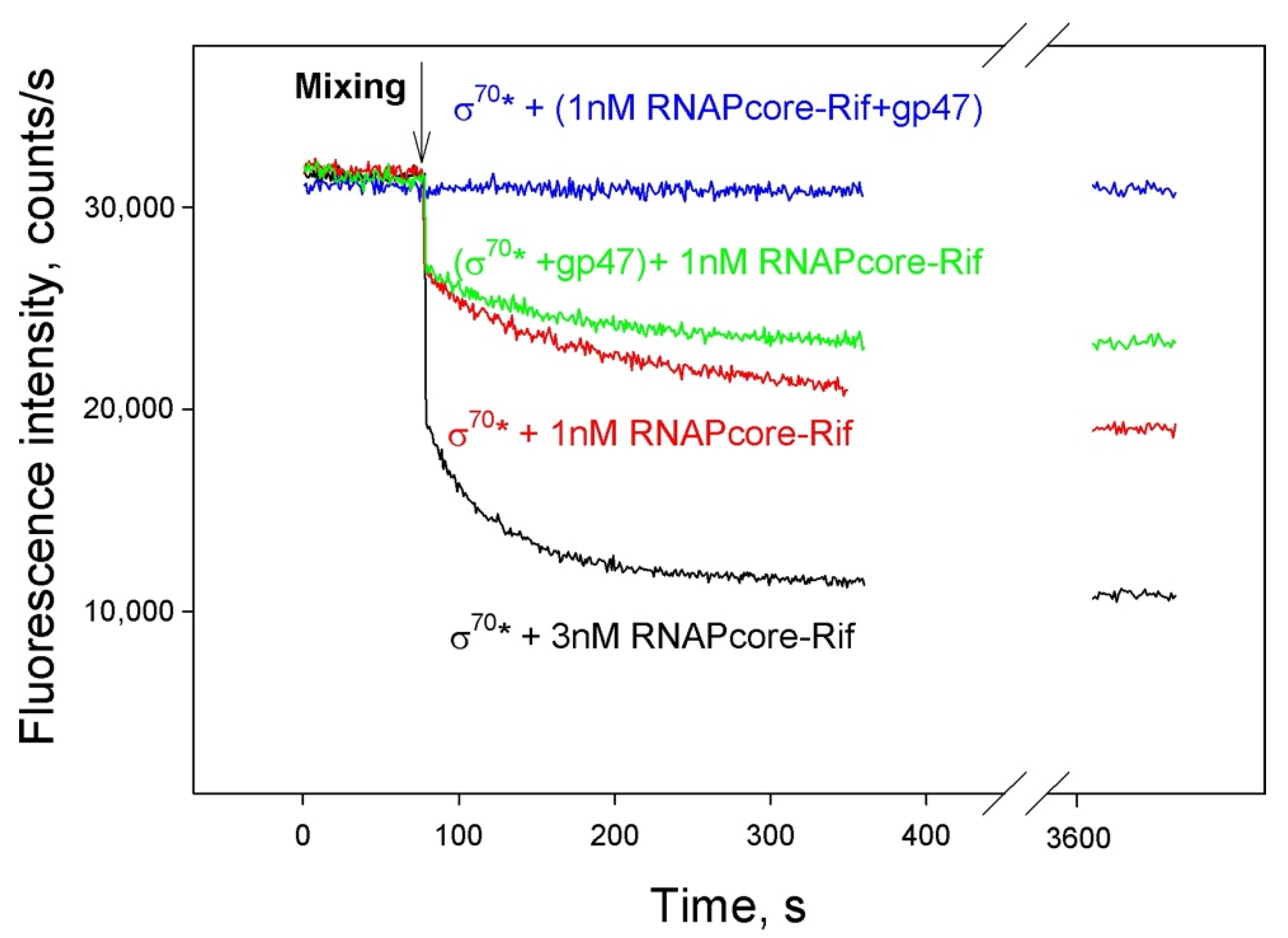
3.4. SaPh711_gp47 Sigma Factor Forms a Stable Complex with Host Core RNAP
3.5. Identification of 7-11 σ70 -Dependent Promoters
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kropinski, A.M.; Lingohr, E.J.; Ackermann, H.W. The genome sequence of enterobacterial phage 7–11, which possesses an unusually elongated head. Arch. Virol. 2010, 156, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Savalia, D.; Westblade, L.F.; Goel, M.; Florens, L.; Kemp, P.; Akulenko, N.; Pavlova, O.; Padovan, J.C.; Chait, B.T.; Washburn, M.P.; et al. Genomic and Proteomic Analysis of phiEco32, a Novel Escherichia coli Phage. J. Mol. Biol. 2008, 377, 774–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, M.K.; Eriksson, H.; Kasuga, K.; Haggård-Ljungquist, E.; Nilsson, A.S. Genomic, Proteomic, Morphological, and Phylogenetic Analyses of vB_EcoP_SU10, a Podoviridae Phage with C3 Morphology. PLoS ONE 2014, 9, e116294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Chen, M.; Tang, F.; Yao, H.; Lu, C.; Zhang, W. Complete Genome Sequence of the Novel Lytic Avian Pathogenic Coliphage NJ01. J. Virol. 2012, 86, 13874–13875. [Google Scholar] [CrossRef] [Green Version]
- Nho, S.-W.; Ha, M.-A.; Kim, K.-S.; Kim, T.-H.; Jang, H.-B.; Cha, I.-S.; Park, S.-B.; Kim, Y.-K.; Jung, T.-S. Complete Genome Sequence of the Bacteriophages ECBP1 and ECBP2 Isolated from Two Different Escherichia coli Strains. J. Virol. 2012, 86, 12439–12440. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, O.; Lavysh, D.; Klimuk, E.; Djordjevic, M.; Ravcheev, D.A.; Gelfand, M.S.; Severinov, K.; Akulenko, N. Temporal regulation of gene expression of the Escherichia coli bacteriophage phiEco32. J. Mol. Biol. 2012, 416, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Klimuk, E.; Akulenko, N.; Makarova, K.S.; Ceyssens, P.J.; Volchenkov, I.; Lavigne, R.; Severinov, K. Host RNA polymerase inhibitors encoded by φKMV-like phages of pseudomonas. Virology 2013, 436, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Klimuk, E.; Mekler, V.; Lavysh, D.; Serebryakova, M.; Akulenko, N.; Severinov, K. Novel Escherichia coli RNA Polymerase Binding Protein Encoded by Bacteriophage T5. Viruses 2020, 12, 807. [Google Scholar] [CrossRef]
- Mironov, A.A.; Vinokurova, N.P.; Gel’fand, M.S. Software for analyzing bacterial genomes. Mol. Biol. 2000, 34, 253–262. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989; ISBN 978-0879693091. [Google Scholar]
- Decker, K.; Krauel, V.; Meesmann, A.; Heller, K.J. Lytic conversion of Escherichia coli by bacteriophage T5: Blocking of the FhuA receptor protein by a lipoprotein expressed early during infection. Mol. Microbiol. 1994, 12, 321–332. [Google Scholar] [CrossRef]
- Kashlev, M.; Nudler, E.; Severinov, K.; Borukhov, S.; Komissarova, N.; Goldfarb, A. Histidine-tagged RNA polymerase of Escherichia coli and transcription in solid phase. Methods Enzymol. 1996, 274, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Severinov, K.; Darst, S.A. A mutant RNA polymerase that forms unusual open promoter complexes. Proc. Natl. Acad. Sci. USA 1997, 94, 13481–13486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frange, P.; Leruez-Ville, M. Maribavir, brincidofovir and letermovir: Efficacy and safety of new antiviral drugs for treating cytomegalovirus infections. Médecine Mal. Infect. 2018, 48, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Klimuk, E.; Bogdanova, E.; Nagornykh, M.; Rodic, A.; Djordjevic, M.; Medvedeva, S.; Pavlova, O.; Severinov, K. Controller protein of restriction–modification system Kpn2I affects transcription of its gene by acting as a transcription elongation roadblock. Nucleic Acids Res. 2018, 46, 10810–10826. [Google Scholar] [CrossRef] [PubMed]
- Mekler, V.; Severinov, K. RNA polymerase molecular beacon as tool for studies of RNA polymerase-promoter interactions. Methods 2015, 86, 19–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knight, J.L.; Mekler, V.; Mukhopadhyay, J.; Ebright, R.H.; Levy, R.M. Distance-Restrained Docking of Rifampicin and Rifamycin SV to RNA Polymerase Using Systematic FRET Measurements: Developing Benchmarks of Model Quality and Reliability. Biophys. J. 2005, 88, 925–938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, E.; Liu, M.; Sheppard, C.; Mekler, V.; Cámara, B.; Liu, B.; Simpson, P.; Cota, E.; Severinov, K.; Matthews, S.; et al. Structural and Mechanistic Basis for the Inhibition of Escherichia coli RNA Polymerase by T7 Gp2. Mol. Cell 2012, 47, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Krisch, H.M.; Comeau, A.M. The immense journey of bacteriophage T4—From d’Hérelle to Delbrück and then to Darwin and beyond. Res. Microbiol. 2008, 159, 314–324. [Google Scholar] [CrossRef]
- Abbasifar, R.; Griffiths, M.W.; Sabour, P.M.; Ackermann, H.W.; Vandersteegen, K.; Lavigne, R.; Noben, J.P.; Alanis Villa, A.; Abbasifar, A.; Nash, J.H.E.; et al. Supersize me: Cronobacter sakazakii phage GAP32. Virology 2014, 460, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME Suite: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Münch, R.; Hiller, K.; Barg, H.; Heldt, D.; Linz, S.; Wingender, E.; Jahn, D. PRODORIC: Prokaryotic database of gene regulation. Nucleic Acids Res. 2003, 31, 266–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kadesch, T.R.; Rosenberg, S.; Chamberlin, M.J. Binding of Escherichia coli RNA Polymerase Holoenzyme to Bacteriophage T7 DNA Measurements of Binding at Bacteriophage T7 Promoter A1 Using a Template Competition Assay. J. Mol. Biol. 1982, 155, 1–29. [Google Scholar] [CrossRef]
- Hawley, D.K.; Mcclure, W.R. Compilation and analysis of Escherichia coli promoter DNA sequences. Nucleic Acids Res. 1983, 11, 2237–2255. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Tuerk, C.; Gauss, P.; Thermes, C.; Groebe, D.R.; Gayle, M.; Guild, N.; Stormo, G.; d’Aubenton-Carafa, Y.; Uhlenbeck, O.C.; Tinoco, I. CUUCGG hairpins: Extraordinarily stable RNA secondary structures associated with various biochemical processes. Proc. Natl. Acad. Sci. USA 1988, 85, 1364–1368. [Google Scholar] [CrossRef] [Green Version]
- Gorski, K.; Roch, J.M.; Prentki, P.; Krisch, H.M. The stability of bacteriophage T4 gene 32 mRNA: A 5’ leader sequence that can stabilize mRNA transcripts. Cell 1985, 43, 461–469. [Google Scholar] [CrossRef]
- Loayza, D.; Carpousis, A.J.; Krisch, H.M. Gene 32 transcription and mRNA processing in T4-related bacteriophages. Mol. Microbiol. 1991, 5, 715–725. [Google Scholar] [CrossRef]
- Vandenberghe, A.; Min Jou, W.; Fiers, W. 3’-Terminal nucletide sequence (n equals 361) of bacteriophage MS2 RNA. Proc. Natl. Acad. Sci. USA 1975, 72, 2559–2562. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lavysh, D.; Mekler, V.; Klimuk, E.; Severinov, K. Regulation of Gene Expression of phiEco32-like Bacteriophage 7-11. Viruses 2022, 14, 555. https://doi.org/10.3390/v14030555
Lavysh D, Mekler V, Klimuk E, Severinov K. Regulation of Gene Expression of phiEco32-like Bacteriophage 7-11. Viruses. 2022; 14(3):555. https://doi.org/10.3390/v14030555
Chicago/Turabian StyleLavysh, Daria, Vladimir Mekler, Evgeny Klimuk, and Konstantin Severinov. 2022. "Regulation of Gene Expression of phiEco32-like Bacteriophage 7-11" Viruses 14, no. 3: 555. https://doi.org/10.3390/v14030555