
Genomic Bootstrap Barcodes and Their Application to Study the Evolution of Sarbecoviruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. DNA Alignment of Genomic Sequences
2.2. Construction of Genomic Bootstrap Barcodes
2.3. SuperTRI Analyses
3. Results
3.1. Phylogenetic Signal in Windows Moving along the Alignment of Sarbecovirus Genomes
3.2. Topological Congruence between the Four SuperTRI Bootstrap Consensus Trees
3.3. Comparison between GB Barcodes Constructed Using Four Sliding Window Sizes
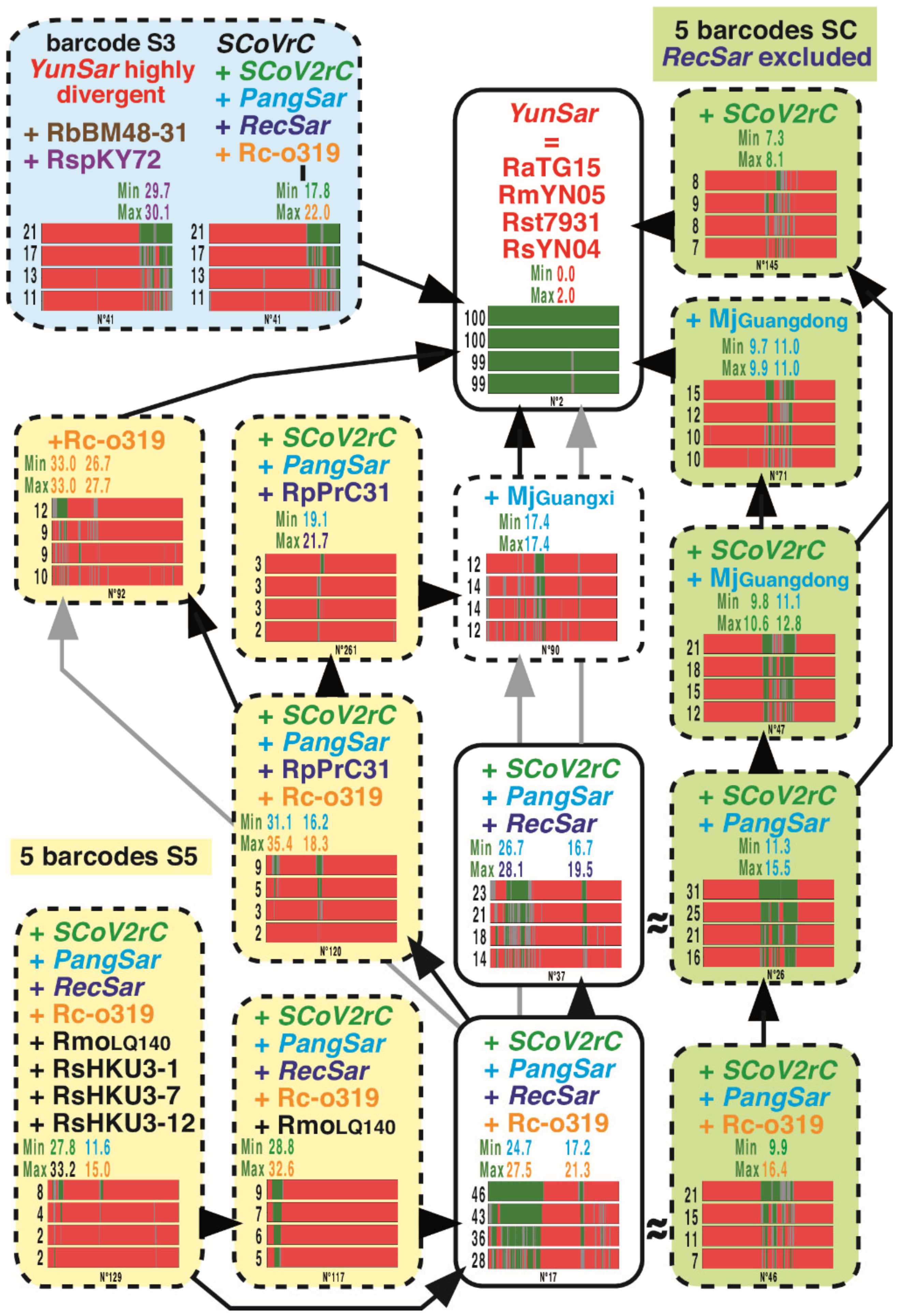
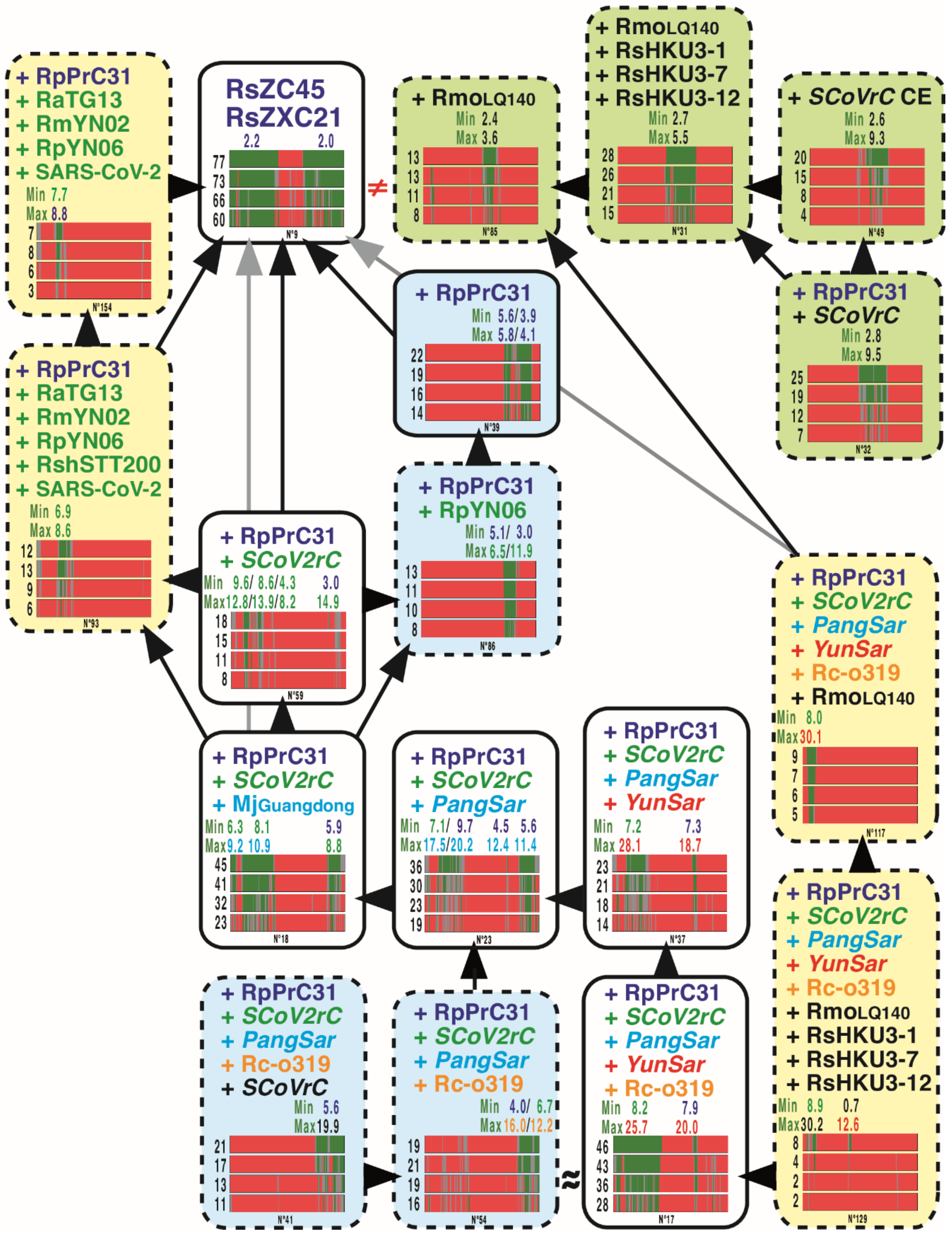
3.4. GB Barcodes of Bipartitions Including YunSar Viruses
3.5. GB Barcodes of Bipartitions Including RecSar Viruses
3.6. GB Barcodes of Bipartitions Containing SARS-CoV-2
3.7. Length Estimation of GRPSs Involving Viruses of the SCoV2rC s.l. Lineage
4. Discussion
4.1. Further Evidence for Four Divergent Sarbecovirus Lineages in Asia
4.2. RdRp Selection of Recombinant Genomes in Bat Reservoirs
4.3. SARS-CoV-2 Is a Mosaic Genome Closely Related to Bat Viruses from Yunnan
4.4. Are CircRNAs Involved in Homologous Recombination?
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Simon-Loriere, E.; Holmes, E.C. Why do RNA viruses recombine? Nat. Rev. Genet. 2011, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Palero, F.; González-Candelas, F. Recombination in viruses: Mechanisms, methods of study, and evolutionary consequences. Infect. Genet. Evol. 2015, 30, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Boni, M.F.; Lemey, P.; Jiang, X.; Lam, T.T.-Y.; Perry, B.W.; Castoe, T.A.; Rambaut, A.; Robertson, D.L. Evolutionary origins of the SARS-CoV-2 sarbecovirus lineage responsible for the COVID-19 pandemic. Nat. Microbiol. 2020, 5, 1408–1417. [Google Scholar] [CrossRef] [PubMed]
- Forni, D.; Cagliani, R.; Sironi, M. Recombination and Positive Selection Differentially Shaped the Diversity of Betacoronavirus Subgenera. Viruses 2020, 12, 1313. [Google Scholar] [CrossRef]
- Hassanin, A.; Grandcolas, P.; Veron, G. COVID-19: Natural or anthropic origin? Mammalia 2021, 85, 1–7. [Google Scholar] [CrossRef]
- Hassanin, A.; Tu, V.T.; Curaudeau, M.; Csorba, G. Inferring the ecological niche of bat viruses closely related to SARS-CoV-2 using phylogeographic analyses of Rhinolophus species. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Wu, Z.; Han, Y.; Wang, Y.; Liu, B.; Zhao, L.; Zhang, J.; Su, H.-X.; Zhao, W.; Liu, L.; Bai, S.; et al. A comprehensive survey of bat sarbecoviruses across China for the origin tracing of SARS-CoV and SARSCoV-2. Res. Sq. 2021. in review. [Google Scholar] [CrossRef]
- Delaune, D.; Hul, V.; Karlsson, E.A.; Hassanin, A.; Ou, T.P.; Baidaliuk, A.; Gámbaro, F.; Prot, M.; Tu, V.T.; Chea, S.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021, 12, 1–7. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Ni Chia, W.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Murakami, S.; Kitamura, T.; Suzuki, J.; Sato, R.; Aoi, T.; Fujii, M.; Matsugo, H.; Kamiki, H.; Ishida, H.; Takenaka-Uema, A.; et al. Detection and Characterization of Bat Sarbecovirus Phylogenetically Related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 2020, 26, 3025–3029. [Google Scholar] [CrossRef]
- Drexler, J.F.; Gloza-Rausch, F.; Glende, J.; Corman, V.M.; Muth, D.; Goettsche, M.; Seebens, A.; Niedrig, M.; Pfefferle, S.; Yordanov, S.; et al. Genomic Characterization of Severe Acute Respiratory Syndrome-Related Coronavirus in European Bats and Classification of Coronaviruses Based on Partial RNA-Dependent RNA Polymerase Gene Sequences. J. Virol. 2010, 84, 11336–11349. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Tong, S. Complete Genome Sequence of a Severe Acute Respiratory Syndrome-Related Coronavirus from Kenyan Bats. Microbiol. Resour. Announc. 2019, 8, e00548-19. [Google Scholar] [CrossRef]
- IUCN. IUCN Red List of Threatened Species; IUCN: Gland, Switzerland, 2008; Available online: http://www.iucnredlist.org (accessed on 27 August 2021).
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 2021, 184, 4380–4391. [Google Scholar] [CrossRef] [PubMed]
- Hassanin, A. Variation in synonymous nucleotide composition among genomes of sarbecoviruses and consequences for the origin of COVID-19. bioRxiv 2021. in review. [Google Scholar] [CrossRef]
- Guo, H.; Hu, B.; Si, H.-R.; Zhu, Y.; Zhang, W.; Li, B.; Li, A.; Geng, R.; Lin, H.-F.; Yang, X.-L.; et al. Identification of a novel lineage bat SARS-related coronaviruses that use bat ACE2 receptor. Emerg. Microbes Infect. 2021, 10, 1507–1514. [Google Scholar] [CrossRef]
- Ropiquet, A.; Li, B.; Hassanin, A. SuperTRI: A new approach based on branch support analyses of multiple independent data sets for assessing reliability of phylogenetic inferences. Comptes Rendus. Biol. 2009, 332, 832–847. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Felsenstein, J. Confidence limits on phylogenies: An approach using the bootstrap. Evolution 1985, 39, 783–791. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (and Other Methods), Version 4; Sinauer Associates: Sunderland, MA, USA, 2003. [Google Scholar]
- Koçhan, N.; Eskier, D.; Suner, A.; Karakülah, G.; Oktay, Y. Different selection dynamics of S and RdRp between SARS-CoV-2 genomes with and without the dominant mutations. Infect. Genet. Evol. 2021, 91, 104796. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Zhu, C.; Ai, L.; He, T.; Wang, Y.; Ye, F.; Yang, L.; Ding, C.; Zhu, X.; Lv, R.; et al. Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerg. Microbes Infect. 2018, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-L.; Wang, J.-L.; Ma, X.-H.; Sun, X.-M.; Li, J.-S.; Yang, X.-F.; Shi, W.-F.; Duan, Z.-J. A novel SARS-CoV-2 related coronavirus with complex recombination isolated from bats in Yunnan province, China. Emerg. Microbes Infect. 2021, 10, 1683–1690. [Google Scholar] [CrossRef]
- Hon, C.-C.; Lam, T.Y.; Shi, Z.-L.; Drummond, A.; Yip, C.-W.; Zeng, F.; Lam, P.-Y.; Leung, F.C.-C. Evidence of the Recombinant Origin of a Bat Severe Acute Respiratory Syndrome (SARS)-Like Coronavirus and Its Implications on the Direct Ancestor of SARS Coronavirus. J. Virol. 2008, 82, 1819–1826. [Google Scholar] [CrossRef]
- Lam, T.T.-Y.; Jia, N.; Zhang, Y.-W.; Shum, M.H.-H.; Jiang, J.-F.; Zhu, H.-C.; Tong, Y.-G.; Shi, Y.-X.; Ni, X.-B.; Liao, Y.-S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. Automated Phylogenetic Detection of Recombination Using a Genetic Algorithm. Mol. Biol. Evol. 2006, 23, 1891–1901. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Segreto, R.; Deigin, Y. The genetic structure of SARS-CoV-2 does not rule out a laboratory origin. BioEssays 2021, 43, e2000240. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Chen, W.; Chen, J.-P. Viral Metagenomics Revealed Sendai Virus and Coronavirus Infection of Malayan Pangolins (Manis javanica). Viruses 2019, 11, 979. [Google Scholar] [CrossRef] [PubMed]
- Temmam, S.; Vongphayloth, K.; Salazar, E.B.; Munier, S.; Bonomi, M.; Régnault, B.; Douangboubpha, B.; Karami, Y.; Chretien, D.; Sanamxay, D.; et al. Coronaviruses with a SARS-CoV-2-like receptor-binding do-main allowing ACE2-mediated entry into human cells isolated from bats of Indochinese peninsula. Res. Sq. 2021. in review. [Google Scholar] [CrossRef]
- Johnson, B.A.; Xie, X.; Bailey, A.L.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; et al. Loss of furin cleavage site attenuates SARS-CoV-2 pathogenesis. Nature 2021, 591, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]
- Chen, L.-L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Xie, R.; Zhang, Y.; Zhang, J.; Li, J.; Zhou, X. The Role of Circular RNAs in Immune-Related Diseases. Front. Immunol. 2020, 11, 545. [Google Scholar] [CrossRef]
- Toptan, T.; Abere, B.; Nalesnik, M.A.; Swerdlow, S.H.; Ranganathan, S.; Lee, N.; Shair, K.H.Y.; Moore, P.S.; Chang, Y. Circular DNA tumor viruses make circular RNAs. Proc. Natl. Acad. Sci. USA 2018, 115, E8737–E8745. [Google Scholar] [CrossRef]
- Ungerleider, N.; Concha, M.; Lin, Z.; Roberts, C.; Wang, X.; Cao, S.; Baddoo, M.; Moss, W.N.; Yu, Y.; Seddon, M.; et al. The Epstein Barr virus circRNAome. PLoS Pathog. 2018, 14, e1007206. [Google Scholar] [CrossRef]
- Hassanin, A.; Jones, H.; Ropiquet, A. SARS-CoV-2-like Viruses from Captive Guangdong Pangolins Generate Circular RNAs. 2020. Available online: https://hal.archives-ouvertes.fr/hal-02616966 (accessed on 27 August 2021).
- Cai, Z.; Lu, C.; He, J.; Liu, L.; Zou, Y.; Zhang, Z.; Zhu, Z.; Ge, X.; Wu, A.; Jiang, T.; et al. Identification and characterization of circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2. Brief. Bioinform. 2021, 22, 1297–1308. [Google Scholar] [CrossRef]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Mac Kain, A.; Joffret, M.-L.; Delpeyroux, F.; Vignuzzi, M.; Bessaud, M. A cold case: Non-replicative recombination in positive-strand RNA viruses. Virologie 2021, 25, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Alhasan, A.A.; Izuogu, O.G.; Al-Balool, H.H.; Steyn, J.S.; Evans, A.; Colzani, M.; Ghevaert, C.; Mountford, J.C.; Marenah, L.; Elliott, D.; et al. Circular RNA enrichment in platelets is a signature of transcriptome degradation. Blood 2016, 127, e1–e11. [Google Scholar] [CrossRef] [PubMed]
- Bera, S.C.; Seifert, M.; Kirchdoerfer, R.N.; van Nies, P.; Wubulikasimu, Y.; Quack, S.; Papini, F.S.; Arnold, J.J.; Canard, B.; Cameron, C.E.; et al. The nucleotide addition cycle of the SARS-CoV-2 polymerase. Cell Rep. 2021, 36, 109650. [Google Scholar] [CrossRef]
- Chrisman, B.S.; Paskov, K.; Stockham, N.; Tabatabaei, K.; Jung, J.-Y.; Washington, P.; Varma, M.; Sun, M.W.; Maleki, S.; Wall, D.P. Indels in SARS-CoV-2 occur at template-switching hotspots. BioData Min. 2021, 14, 1–16. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanin, A.; Rambaud, O.; Klein, D. Genomic Bootstrap Barcodes and Their Application to Study the Evolution of Sarbecoviruses. Viruses 2022, 14, 440. https://doi.org/10.3390/v14020440
Hassanin A, Rambaud O, Klein D. Genomic Bootstrap Barcodes and Their Application to Study the Evolution of Sarbecoviruses. Viruses. 2022; 14(2):440. https://doi.org/10.3390/v14020440
Chicago/Turabian StyleHassanin, Alexandre, Opale Rambaud, and Dylan Klein. 2022. "Genomic Bootstrap Barcodes and Their Application to Study the Evolution of Sarbecoviruses" Viruses 14, no. 2: 440. https://doi.org/10.3390/v14020440