
Characterization of Near Full-Length Transmitted/Founder HIV-1 Subtype D and A/D Recombinant Genomes in a Heterosexual Ugandan Population (2006–2011)

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. HIV-1 Near Full-Length and Half Genomes
2.2.1. Amplification of Near Full-Length Genomes
2.2.2. HIV-1 Half Genomes
2.2.3. SMRT Sequencing of HIV-1 Genomes to Identify T/F
2.2.4. Subtype Classification
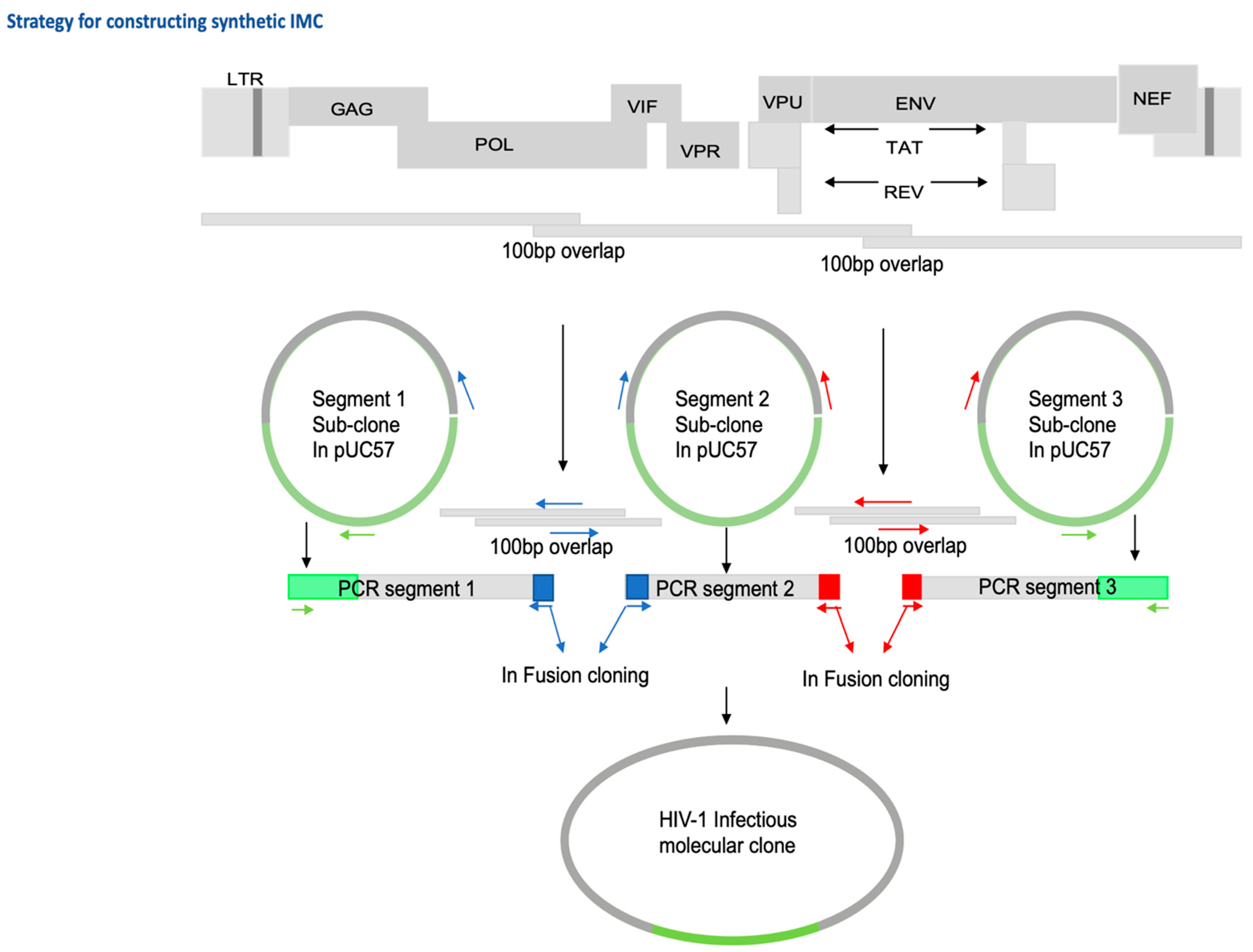
2.3. Generation of Infectious Molecular Clones and In-Fusion Cloning
2.4. Generation of Virus Stocks and Determination of Viral Replicative Capacity
2.5. Statistical Analyses
3. Results
3.1. Study Subjects
3.2. HIV-1 Genetic Diversity
3.3. Model Predictions Analysis
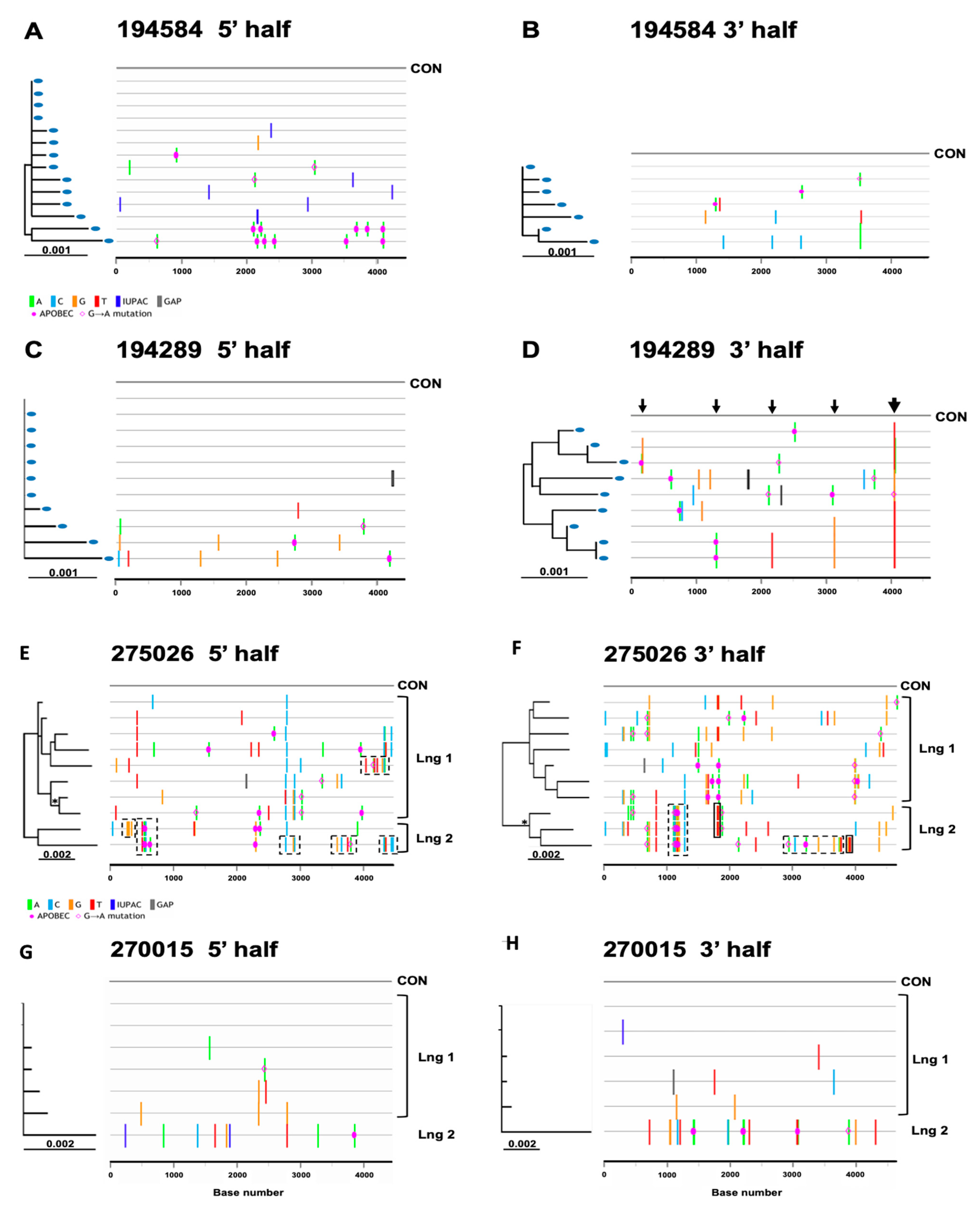
3.4. Inference of Transmitted/Founder Viruses
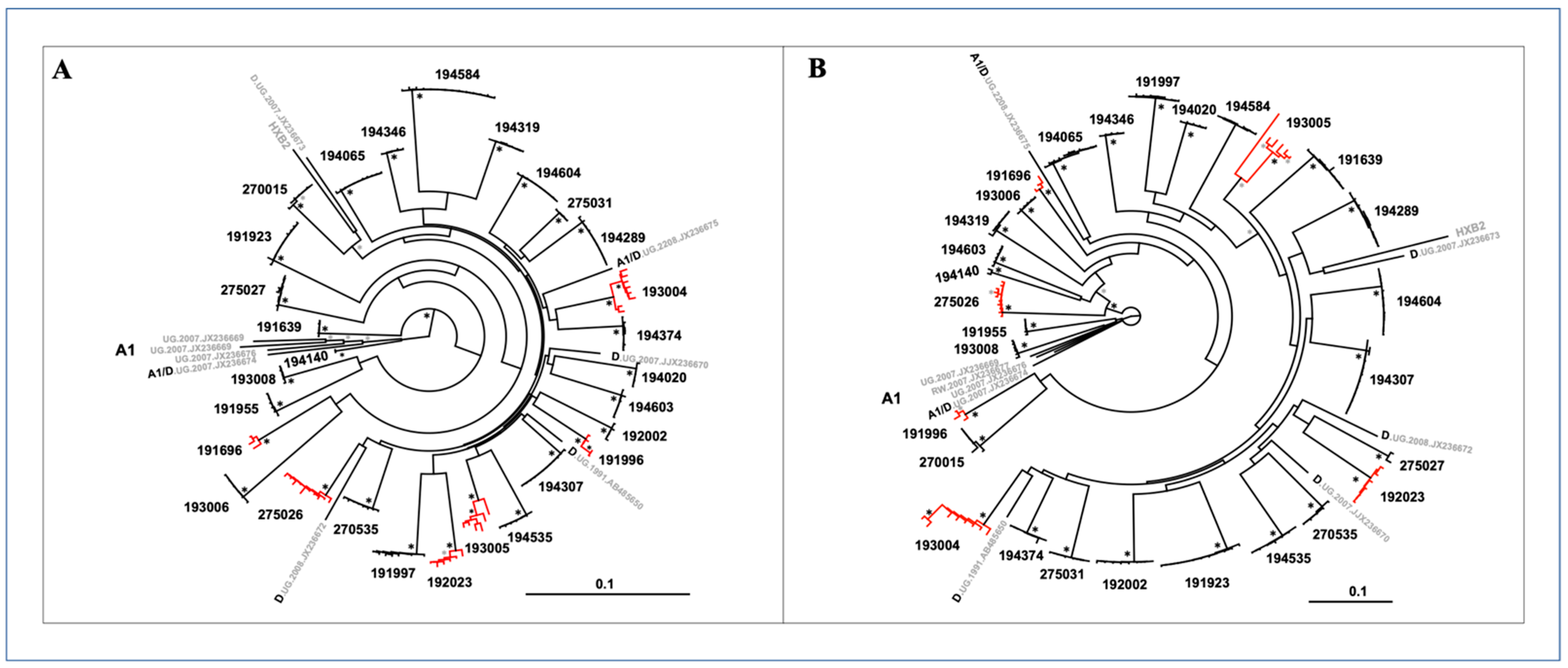
3.5. Subtype Composition
3.6. Recombination Breakpoints
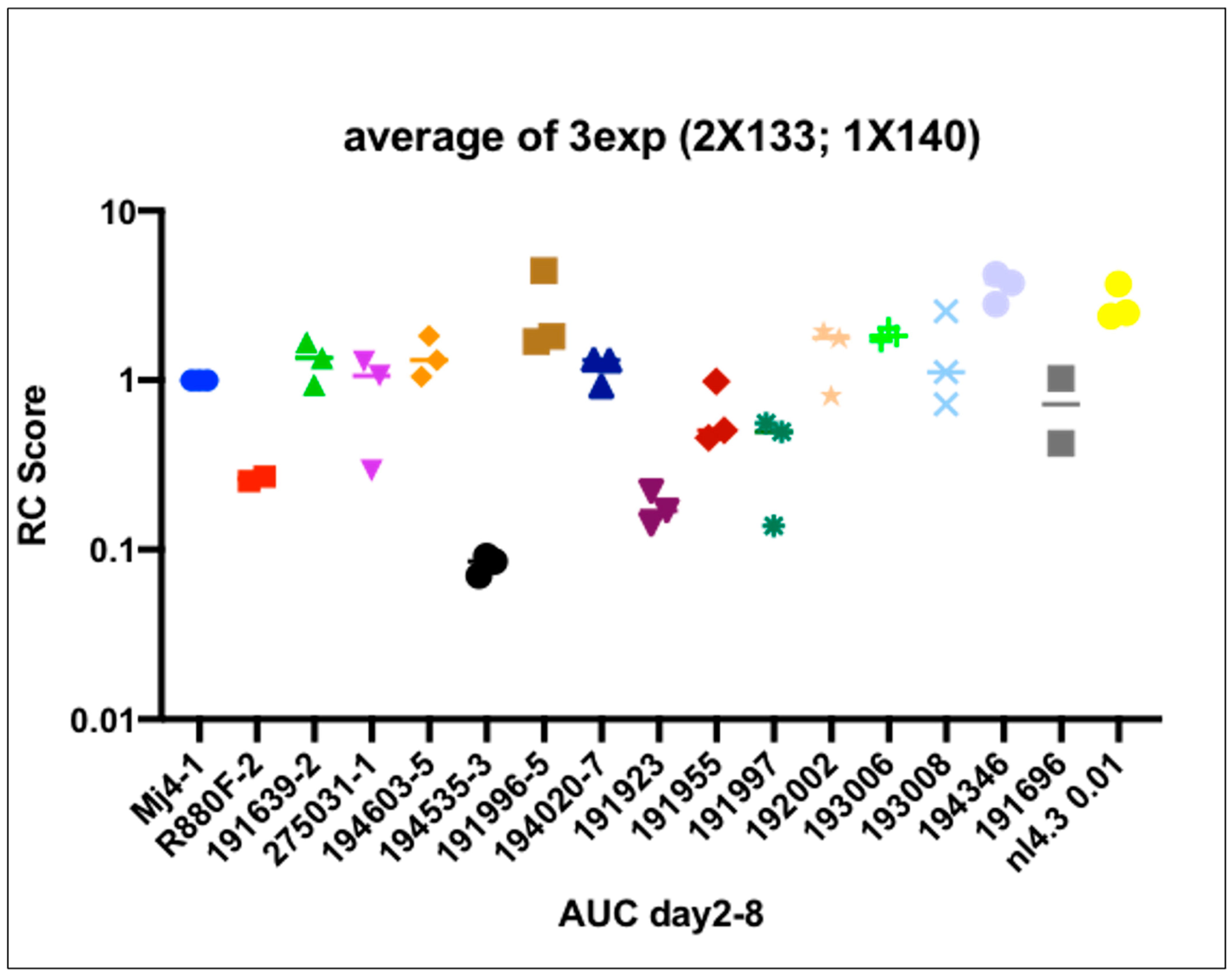
3.7. Viral Replicative Capacity
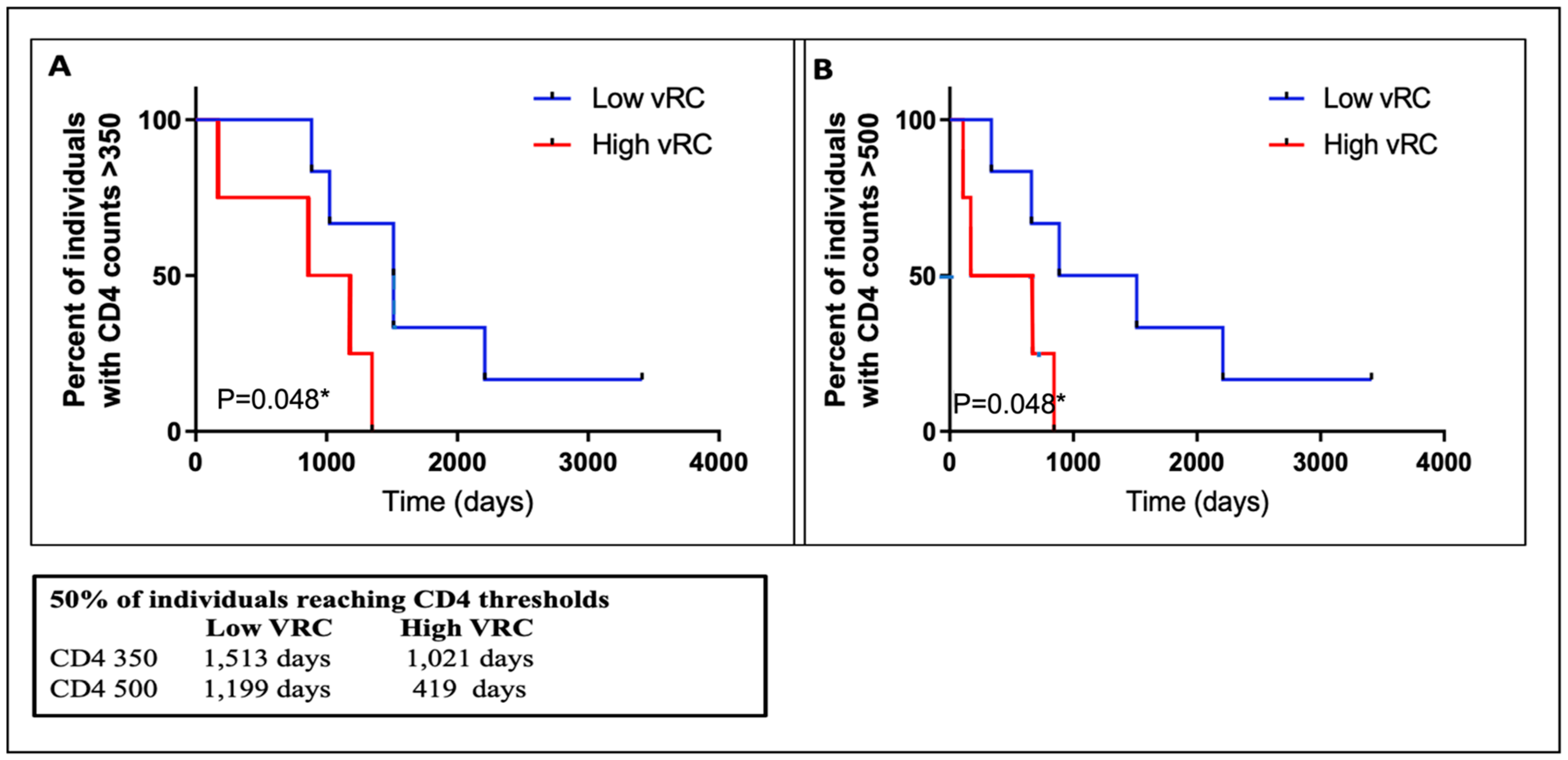
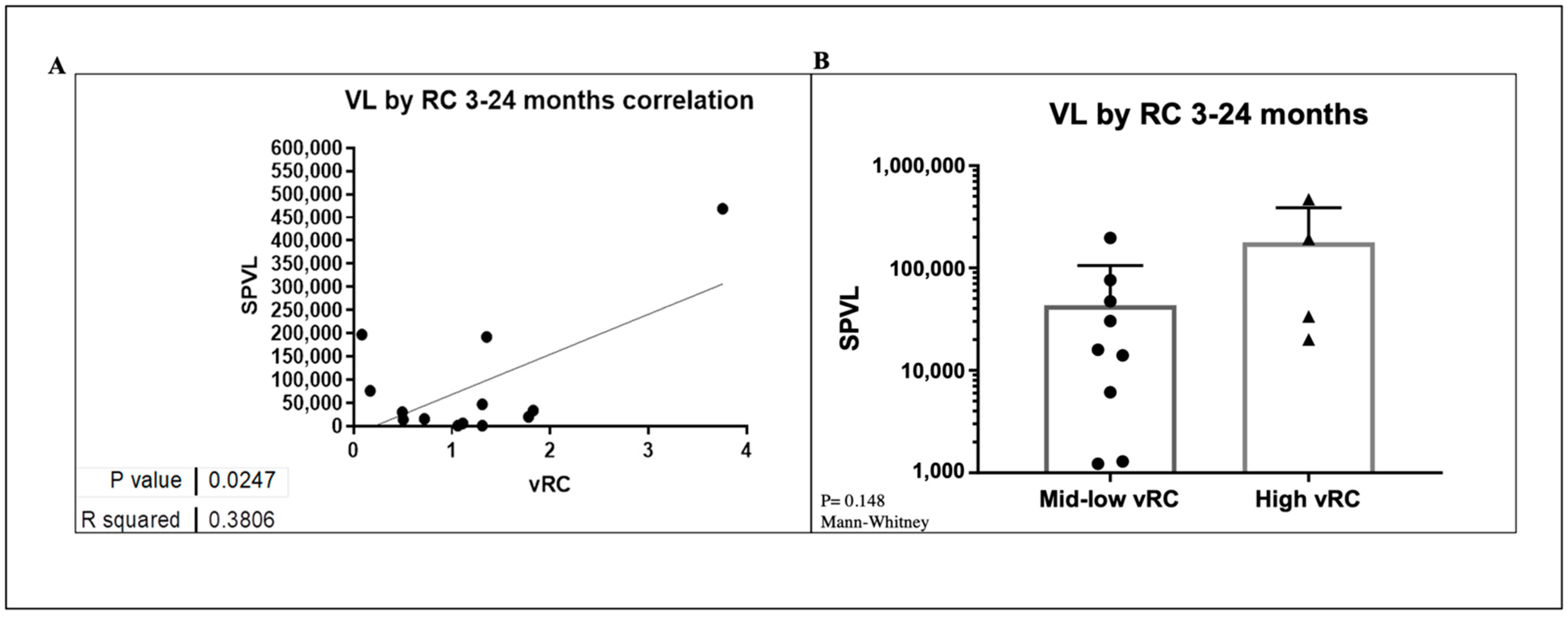
3.8. VRC Association with CD4+T Cell Count and Set Point Viral Load
3.9. Amino Acid Signatures of T/F Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- UNAIDS. UNAIDS Data 2019. Joint United Nations Programme on HIV/AIDS. Available online: https://www.unaids.org/sites/default/files/media_asset/2019-UNAIDS-data_en.pdf (accessed on 13 February 2020).
- Lihana, R.W.; Ssemwanga, D.; Abimiku, A.; Ndembi, N. Update on HIV-1 diversity in Africa: A decade in review. Aids Rev. 2012, 14, 83–100. [Google Scholar]
- Yirrell, D.L.; Kaleebu, P.; Morgan, D.; Watera, C.; Magambo, B.; Lyagoba, F.; Whitworth, J. Inter- and intra-genic intersubtype HIV-1 recombination in rural and semi-urban Uganda. AIDS 2002, 16, 279–286. [Google Scholar] [CrossRef]
- Ssemwanga, D.; Bbosa, N.; Nsubuga, R.; Ssekagiri, A.; Kapaata, A.; Nannyonjo, M.; Nassolo, F.; Karabarinde, A.; Mugisha, J.; Seeley, J.; et al. The Molecular Epidemiology and Transmission Dynamics of HIV Type 1 in a General Population Cohort in Uganda. Viruses 2020, 12, 1283. [Google Scholar] [CrossRef]
- Eshleman, S.H.; Gonzales, M.J.; Becker-Pergola, G.; Cunningham, S.C.; Guay, L.A.; Jackson, J.B.; Shafer, R.W. Identification of Ugandan HIV Type 1 Variants with Unique Patterns of Recombination in pol Involving Subtypes A and D. AIDS Res. Hum. Retrovir. 2002, 18, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-Dörner, L.; Bonsall, D.; Hoppe, A.; Brown, A.L.; PANGEA Consortium and Rakai Health Sciences Program; et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef] [Green Version]
- Ratmann, O.; Kagaayi, J.; Hall, M.; Golubchick, T.; Kigozi, G.; Xi, X.; Wymant, C.; Nakigozi, G.; Abeler-Dörner, L.; Bonsall, D.; et al. Quantifying HIV transmission flow between high-prevalence hotspots and surrounding communities: A population-based study in Rakai, Uganda. Lancet HIV 2020, 7, e173–e183. [Google Scholar] [CrossRef] [Green Version]
- Harris, M.E.; Serwadda, D.; Sewankambo, N.; Kim, B.; Kigozi, G.; Kiwanuka, N.; Phillips, J.B.; Wabwire, F.; Meehen, M.; Lutalo, T.; et al. Among 46 Near Full Length HIV Type 1 Genome Sequences from Rakai District, Uganda, Subtype D and AD Recombinants Predominate. AIDS Res. Hum. Retrovir. 2002, 18, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.Q.; Bangsberg, D.R.; Mo, T.; Lachowski, C.; Brumme, C.J.; Zhang, W.; Lima, V.D.; Boum, Y., II; Mwebesa, B.B.; Muzoora, C.; et al. Prevalence and clinical impacts of HIV-1 intersubtype recom-binants in Uganda revealed by near-full-genome population and deep sequencing approaches. AIDS 2017, 31, 2345. [Google Scholar] [CrossRef]
- Grant, H.E.; Hodcroft, E.B.; Ssemwanga, D.; Kitayimbwa, J.; Yebra, G.; Gomez, L.R.E.; Frampton, D.; Gall, A.; Kellam, P.; De Oliveira, T.; et al. Pervasive and non-random recombination in near full-length HIV genomes from Uganda. Virus Evol. 2020, 6, veaa004. [Google Scholar] [CrossRef]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, M.-R.; Anderson, J.A.; Giorgi, E.; Seoighe, C.; Mlisana, K.; Ping, L.-H. Quantitating the multiplicity of infection with human immunodeficiency virus type 1 subtype C reveals a non-poisson distribution of transmitted variants. J. Virol. 2009, 83, 3556–3567. [Google Scholar] [CrossRef] [Green Version]
- Deymier, M.J.; Ende, Z.; Fenton-May, A.; Dilernia, D.A.; Kilembe, W.; Allen, S.A.; Borrow, P.; Hunter, E. Heterosexual Transmission of Subtype C HIV-1 Selects Consensus-Like Variants without Increased Replicative Capacity or Interferon-α Resistance. PLoS Pathog. 2015, 11, e1005154. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.S.; Bibollet-Ruche, F.; Sherrill-Mix, S.; Learn, G.H.; Plenderleith, L.; Smith, A.G.; Barbian, H.J.; Russell, R.M.; Gondim, M.V.P.; Bahari, C.Y.; et al. Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl. Acad. Sci. USA 2017, 114, E590–E599. [Google Scholar] [CrossRef] [Green Version]
- Baalwa, J.; Wang, S.; Parrish, N.F.; Decker, J.M.; Keele, B.F.; Learn, G.H.; Yue, L.; Ruzagira, E.; Ssemwanga, D.; Kamali, A.; et al. Molecular identification, cloning and characterization of transmit-ted/founder HIV-1 subtype A, D and A/D infectious molecular clones. Virology 2013, 436, 33–48. [Google Scholar] [CrossRef] [Green Version]
- Price, A.M.; Kilembe, W.; Ruzagira, E.; Karita, E.; Inambao, M.; Sanders, E.J.; Anzala, O.; Allen, S.; Edward, A.V.; Kaleebu, P.; et al. Cohort Profile: IAVI’s HIV epidemiology and early infection cohort studies in Africa to support vaccine discovery. Int. J. Epidemiol. 2020, 50, 29–30. [Google Scholar] [CrossRef]
- Deymier, M.J.; Claiborne, D.T.; Ende, Z.; Ratner, H.K.; Kilembe, W.; Allen, S.; Hunter, E. Particle infectivity of HIV-1 full-length genome infectious molecular clones in a subtype C heterosexual transmission pair following high fidelity amplification and unbiased cloning. Virology 2014, 468–470, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Gonzalez, J.F.; Bailes, E.; Pham, K.T.; Salazar, M.G.; Guffey, M.B.; Keele, B.F.; Derdeyn, C.A.; Farmer, P.; Hunter, E.; Allen, S.; et al. Deciphering Human Immunodeficiency Virus Type 1 Transmission and Early Envelope Diversification by Single-Genome Amplification and Sequencing. J. Virol. 2008, 82, 3952–3970. [Google Scholar] [CrossRef] [Green Version]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Dilernia, D.A.; Chien, J.-T.; Monaco, D.C.; Brown, M.P.S.; Ende, Z.; Deymier, M.J.; Yue, L.; Paxinos, E.E.; Allen, S.; Tirado-Ramos, A.; et al. Multiplexed highly-accurate DNA sequencing of close-ly-related HIV-1 variants using continuous long reads from single molecule, real-time sequencing. Nucleic Acids Res. 2015, 43, e129. [Google Scholar] [CrossRef] [Green Version]
- Siepel, A.C.; Halpern, A.L.; Macken, C.; Korber, B. A Computer Program Designed to Screen Rapidly for HIV Type 1 Intersubtype Recombinant Sequences. AIDS Res. Hum. Retrovir. 1995, 11, 1413–1416. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, T.; Deforche, K.; Cassol, S.; Salminen, M.; Paraskevis, D.; Seebregts, C.; Snoeck, J.; Van Rensburg, E.J.; Wensing, A.M.J.; van de Vijver, D.A.; et al. An automated genotyping system for analysis of HIV-1 and other microbial sequences. Bioinformatics 2005, 21, 3797–3800. [Google Scholar] [CrossRef] [Green Version]
- Schultz, A.-K.; Zhang, M.; Bulla, I.; Leitner, T.; Korber, B.; Morgenstern, B. jpHMM: Improving the reliability of recombination prediction in HIV. Nucleic Acids Res. 2009, 37, W647–W651. [Google Scholar] [CrossRef] [Green Version]
- Wright, J.K.; Brumme, Z.L.; Carlson, J.M.; Heckerman, D.; Kadie, C.M.; Brumme, C.J.; Wang, B.; Losina, E.; Miura, T.; Chonco, F.; et al. Gag-Protease-Mediated Replication Capacity in HIV-1 Subtype C Chronic Infection: Associations with HLA Type and Clinical Parameters. J. Virol. 2010, 84, 10820–10831. [Google Scholar] [CrossRef] [Green Version]
- Claiborne, D.T.; Prince, J.L.; Hunter, E. A Restriction Enzyme Based Cloning Method to Assess the In vitro Replication Capacity of HIV-1 Subtype C Gag-MJ4 Chimeric Viruses. J. Vis. Exp. 2014, 90, e51506. [Google Scholar] [CrossRef] [Green Version]
- Claiborne, D.T.; Prince, J.L.; Scully, E.; Macharia, G.; Micci, L.; Lawson, B.; Kopycinski, J.; Deymier, M.J.; Vanderford, T.H.; Nganou-Makamdop, K.; et al. Replicative fitness of transmitted HIV-1 drives acute immune activation, proviral load in memory CD4+ T cells, and disease progression. Proc. Natl. Acad. Sci. USA 2015, 112, E1480–E1489. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Bar, K.J.; Wang, S.; Decker, J.M.; Chen, Y.; Sun, C.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Learn, G.H.; Morgan, C.J.; et al. High Multiplicity Infection by HIV-1 in Men Who Have Sex with Men. PLoS Pathog. 2010, 6, e1000890. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Giorgi, E.E.; Ganusov, V.V.; Cai, F.; Athreya, G.; Yoon, H.; Carja, O.; Hora, B.; Hraber, P.; Romero-Severson, E.; et al. Tracking HIV-1 recombination to resolve its contribution to HIV-1 evolution in natural infection. Nat. Commun. 2018, 9, 1928. [Google Scholar] [CrossRef] [Green Version]
- Turk, G.; Carobene, M.G. Deciphering How HIV-1 Intersubtype Recombination Shapes Viral Fitness and Disease Progression. EBioMedicine 2015, 2, 188–189. [Google Scholar] [CrossRef] [Green Version]
- Goepfert, P.A.; Lumm, W.; Farmer, P.; Matthews, P.; Prendergast, A.; Carlson, J.M.; Derdeyn, C.A.; Tang, J.; Kaslow, R.A.; Bansal, A.; et al. Transmission of HIV-1 Gag immune escape mutations is associated with reduced viral load in linked recipients. J. Exp. Med. 2008, 205, 1009–1017. [Google Scholar] [CrossRef]
- Prince, J.L.; Claiborne, D.T.; Carlson, J.M.; Schaefer, M.; Yu, T.; Lahki, S.; Prentice, H.; Yue, L.; Vishwanathan, S.A.; Kilembe, W.; et al. Role of Transmitted Gag CTL Polymorphisms in Defining Replicative Capacity and Early HIV-1 Pathogenesis. PLoS Pathog. 2012, 8, e1003041. [Google Scholar] [CrossRef] [Green Version]
- Fraser, C.; Lythgoe, K.; Leventhal, G.E.; Shirreff, G.; Hollingsworth, T.D.; Alizon, S.; Bonhoeffer, S. Virulence and Pathogenesis of HIV-1 Infection: An Evolutionary Perspective. Science 2014, 343, 1243727. [Google Scholar] [CrossRef] [Green Version]
- Asmal, M.; Hellmann, I.; Liu, W.; Keele, B.F.; Perelson, A.S.; Bhattacharya, T.; Gnanakaran, S.; Daniels, M.; Haynes, B.F.; Korber, B.; et al. A Signature in HIV-1 Envelope Leader Peptide Associated with Transition from Acute to Chronic Infection Impacts Envelope Processing and Infectivity. PLoS ONE 2011, 6, e23673. [Google Scholar] [CrossRef] [Green Version]
- Gnanakaran, S.; Bhattacharya, T.; Daniels, M.; Keele, B.F.; Hraber, P.; Lapedes, A.S.; Shen, T.; Gaschen, B.; Krishnamoorthy, M.; Li, H.; et al. Recurrent Signature Patterns in HIV-1 B Clade Envelope Glycoproteins Associated with either Early or Chronic Infections. PLoS Pathog. 2011, 7, e1002209. [Google Scholar] [CrossRef]
- Haaland, R.E.; Hawkins, P.A.; Salazar-Gonzalez, J.F.; Johnson, A.; Tichacek, A.; Karita, E.; Manigart, O.; Mulenga, J.; Keele, B.F.; Shaw, G.M.; et al. Inflammatory Genital Infections Mitigate a Severe Genetic Bottleneck in Heterosexual Transmission of Subtype A and C HIV-1. PLoS Pathog. 2009, 5, e1000274. [Google Scholar] [CrossRef] [Green Version]
- Kijak, G.H.; Sanders-Buell, E.; Chenine, A.-L.; Eller, M.A.; Goonetilleke, N.; Thomas, R.; Leviyang, S.; Harbolick, E.A.; Bose, M.; Pham, P.; et al. Rare HIV-1 transmitted/founder lineages identified by deep viral sequencing contribute to rapid shifts in dominant quasispecies during acute and early infection. PLoS Pathog. 2017, 13, e1006510. [Google Scholar]
- Capoferri, A.A.; Lamers, M.S.; Grabowski, M.K.; Rose, R.R.; Wawer, M.J.; Serwadda, D.; Gray, R.H.; Quinn, T.C.; Kigozi, G.; Kagaayi, J.; et al. Recombination Analysis of Near Full-Length HIV-1 Sequences and the Identification of a Potential New Circulating Recombinant Form from Rakai, Uganda. AIDS Res. Hum. Retrovir. 2020, 36, 467–474. [Google Scholar] [CrossRef]
- Binley, J.M.; Wrin, T.; Korber, B.; Zwick, M.B.; Wang, M.; Chappey, C.; Stiegler, G.; Kunert, R.; Zolla-Pazner, S.; Katinger, H.; et al. Comprehensive Cross-Clade Neutralization Analysis of a Panel of Anti-Human Immunodeficiency Virus Type 1 Monoclonal Antibodies. J. Virol. 2004, 78, 13232–13252. [Google Scholar] [CrossRef] [Green Version]
- Simon-Lorière, E.; Galetto, R.; Hamoudi, M.; Archer, J.; Lefeuvre, P.; Martin, D.P.; Robertson, D.L.; Negroni, M. Molecular Mechanisms of Recombination Restriction in the Envelope Gene of the Human Immunodeficiency Virus. PLoS Pathog. 2009, 5, e1000418. [Google Scholar] [CrossRef] [Green Version]
- Purcell, D.F.; A Martin, M. Alternative splicing of human immunodeficiency virus type 1 mRNA modulates viral protein expression, replication, and infectivity. J. Virol. 1993, 67, 6365–6378. [Google Scholar] [CrossRef] [Green Version]
- Tazi, J.; Bakkour, N.; Marchand, V.; Ayadi, L.; Aboufirassi, A.; Branlant, C. Alternative splicing: Regulation of HIV-1 multiplication as a target for therapeutic action. FEBS J. 2010, 277, 867–876. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.; O’Connor, D.; Jing, P.; Dzuris, J.; Mothé, B.R.; Vogel, T.U.; Dunphy, E.J.; Liebl, M.E.; Emerson, C.; Wilson, A.N.; et al. Tat-specific cytotoxic T lymphocytes select for SIV escape variants during resolution of primary viraemia. Nature 2000, 407, 386–390. [Google Scholar] [CrossRef]
- Addo, M.; Altfeld, M.; Rosenberg, E.S.; Eldridge, R.L.; Philips, M.N.; Habeeb, K.; Khatri, A.; Brander, C.; Robbins, G.K.; Mazzara, G.P.; et al. The HIV-1 regulatory proteins Tat and Rev are frequently targeted by cytotoxic T lymphocytes derived from HIV-1-infected individuals. Proc. Natl. Acad. Sci. USA 2001, 98, 1781–1786. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, J.; Mauer, E.J.; Sherer, N.M. HIV-1 Gag, Envelope, and Extracellular Determinants Cooperate To Regulate the Stability and Turnover of Virological Synapses. J. Virol. 2016, 90, 6583–6597. [Google Scholar] [CrossRef] [Green Version]
- Prado, J.G.; Prendergast, A.; Thobakgale-Tshabalala, C.; Molina, C.I.J.; Tudor-Williams, G.; Ndung’U, T.; Walker, B.D.; Goulder, P. Replicative Capacity of Human Immunodeficiency Virus Type 1 Transmitted from Mother to Child Is Associated with Pediatric Disease Progression Rate. J. Virol. 2010, 84, 492–502. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SN | SAMPLE-ID | Sample Date | EDI * | Days Post Infection ***** | Initial Viral Load(Copies/mL) | IMC-VRC Score ** | Gender | Age *** | Subtype (Full Genome) | GenBank No. |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 194065 | 12-Jan-10 | 01-Dec-09 | 42 | 254,000 | N/A **** | M | 41 | A/D | MW006066 |
| 2 | 194289 | 29-Mar-10 | 14-Feb-10 | 43 | 1,394,000 | N/A | F | 24 | D | MW006068 |
| 3 | 193006 | 07-Apr-09 | 15-Feb-08 | 51 | 112,000 | 1.84 | M | 24 | A/D | MW006062 |
| 4 | 194604 | 28-Mar-11 | 12-Feb-11 | 44 | 254,516 | N/A | F | 35 | D | MW006076 |
| 5 | 194603 | 21-Jul-10 | 30-May-10 | 52 | 177,000 | 1.38 | F | 33 | A/D | MW006075 |
| 6 | 191696 | 11-Sep-07 | 23-Jul-07 | 50 | 435,000 | 0.72 | M | 29 | A/D | MW006053 |
| 7 | 194535 | 16-Jun-11 | 30-Apr-11 | 47 | 289,930 | 0.08 | M | 39 | D | MW006073 |
| 8 | 275026 | 10-Sep-08 | 21-Jul-08 | 51 | 412,000 | N/A | F | 21 | A/D | MW006079 |
| 9 | 194319 | 09-Mar-09 | 18-Feb-09 | 19 | 602,000 | N/A | F | 26 | A/D | MW006070 |
| 10 | 192023 | 28-Jan-08 | 06-Dec-07 | 53 | 996,000 | N/A | M | 27 | A/D | MW006059 |
| 11 | 191923 | 24-Jan-08 | 30-Nov-07 | 55 | 198,000 | 0.17 | F | 31 | A/D | MW006054 |
| 12 | 193004 | 30-Oct-07 | 26-Sep-07 | 34 | 6690 | N/A | F | 25 | A/D | MW006060 |
| 13 | 193005 | 06-Nov-07 | 11-Oct-07 | 26 | 90,500 | N/A | M | 22 | D | MW006061 |
| 14 | 194020 | 12-Jun-09 | 07-May-09 | 36 | 77,700 | 1.16 | M | 33 | A/D | MW006064 |
| 15 | 194346 | 31-Mar-09 | 28-Feb-09 | 31 | 259,000 | 3.58 | M | 29 | A/D | MW006071 |
| 16 | 191639 | 03-Apr-08 | 13-Feb-08 | 50 | 102,000 | 1.32 | M | 50 | A/D | MW006052 |
| 17 | 270015 | 19-Dec-06 | 08-Dec-06 | 11 | 19,300 | N/A | M | 58 | A/D | MW006077 |
| 18 | 193008 | 15-Jun-09 | 23-May-09 | 23 | 50,500 | 1.46 | M | 27 | A/D | MW006063 |
| 19 | 191955 | 26-Mar-07 | 03-Mar-07 | 23 | 4190 | 0.64 | F | 39 | A/D | MW006055 |
| 20 | 191997 | 15-Jan-08 | 19-Nov-07 | 57 | 4610 | 0.37 | M | 31 | A/D | MW006057 |
| 21 | 192002 | 24-Jul-07 | 04-Jun-07 | 50 | 2420 | 1.54 | F | 27 | D | MW006058 |
| 22 | 275031 | 05-Jun-09 | 11-May-09 | 25 | 3750 | 0.87 | M | 31 | D | MW006081 |
| 23 | 191996 | 19-Sep-08 | 26-Jul-08 | 55 | 201 | 2.64 | F | 37 | A/D | MW006056 |
| 24 | 194307 | 2-Mar-09 | 3-Feb-09 | 27 | 930,000 | N/A | F | 21 | A/D | MW006068 |
| 25 | 194374 | 04-Feb-10 | 24-Dec-09 | 42 | 43,300 | N/A | M | 33 | D | MW006072 |
| 26 | 194584 | 05-Jul-10 | 03-Jun-10 | 32 | 76,700 | N/A | F | 33 | A/D | MW006079 |
| 27 | 270535 | 17-Dec-08 | 05-Oct-08 | 73 | 51,200 | N/A | M | 31 | D | MW006078 |
| 28 | 194140 | 28-May-09 | 07-Apr-09 | 51 | 45,600 | N/A | F | 26 | A/D | MW006067 |
| 29 | 275027 | 01-Oct-08 | 01-Aug-08 | 61 | 14,700 | N/A | F | 22 | A/D | MW006080 |
| Primer Name | Primer Sequence | Primer Use |
|---|---|---|
| UG1.3′3_PlCb | 5′-ACTACTTAAAGCACTCAAGGCAAGCTTTATTG-3′ | cDNA synthesis for near full-length |
| UGOFM19 | 5′-GCACTCAAGGCAAGCTTTATTGAGGCTTA-3′ | cDNA synthesis for near full-length |
| 1U5Cc | 5′-CCTTGAGTGCTTTAAGTAGTGTGTGCCCGTCTGT-3′ | Near full-length genome PCR |
| UG1.3′3_PlCb | 5′-ACTACTTAAAGCACTCAAGGCAAGCTTTATTG-3′ | Near full-length genome PCR |
| 2U5Cd | 5′-AGTAGTGTGTGCCCGTCTGTTGTGTGACTC-3′ | Near full-length genome PCR |
| UG2.3′3′plCb | 5′-TAAAGCACTCAAGGCAAGCTTTATTGAGGCTTA-3′ | Near full-length genome PCR |
| 5FIV-R1 | 5′-CTYTTTCTCCTGTATGCAGACCCC-3′ | cDNA synthesis for 5′ half genome |
| b5r1 | 5′-CTTGCCACACAATCATCACCTGCCAT-3′ | cDNA synthesis for 5′ half genome |
| 1.R3.B3R | 5′-ACTACTTGAAGCACTCAAGGCAAGCTTTAT-3′ | cDNA synthesis for 3′ half genome |
| RVDA-F1 | 5′-GGGTCTCTCDGTTAGACCAGAT-3′ | 5′ half genome PCR |
| b3F1 | 5′-ACAGCAGTACAAATGGCAGTATT-3′ | 3′ half genome PCR |
| 5FV-R22 | 5′-CCTAGTGGGATGTGTACTTCTGAAC-3′ | 5′ half genome PCR |
| b3f3 | 5′-TGGAAAGGTGAAGGGGCAGTAGTAATAC-3′ | 3′ half genome PCR |
| 2.R3.B6R | 5′-TGAAGCACTCAAGGCAAGCTTTATTGAGGC-3′ | 3′ half genome PCR |
| Example of strain-optimized primers | ||
| con1.rev | 5′-TCTGATGCTTTTTGTCTGGTGT-3′ | Generation of IMC (overlap of Fragments 1 & 2) |
| con1.fwd | 5′-ACACCAGACAAAAAGCATCAGA-3′ | Generation of IMC (overlap of Fragments 1 & 2) |
| 318.con2.fwd | 5′-CCATGTGTAAAGCTAACCCCACTC-3′ | Generation of IMC (overlap of Fragments 2 & 3) |
| 318.con2.rev | 5′-GGTTAGCTTTACACATGGTTTTAGAC-3′ | Generation of IMC overlap of Fragments 2 & 3) |
| AmpR.PvuI.fwd | 5′-CGATCGTTGTCAGAAGTAAGTTGGCCGCAGTGTT-3′ | Generation of IMC (ampR) |
| AmpR.PvuI.rev | 5′-TTCTGACAACGATCGGAGGACCGAAGGAGCTAACCGCTT-3′ | Generation of IMC (ampR) |
| Subject ID | HIV Half Genomes (a) | Total Number HIV Genomes | Minimum nt Length of Viral Genome | Maximum nt Length of Viral Genome | Nucleotide Sequence Diversity (mean %) | Nucleotide Sequence Diversity (Range %) | Number of Genomes Analysed (b) | Mean HD | Max HD | Poisson Estimated Days Since MRCA (c) | Lambda (d) | Goodness of Fit p-Value (e) | HD Fit to Poisson | Star Phylogeny | Deviation from Star Phylogeny | Number of Transmitted Viruses |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 275026 | 5′ | 10 | 4474 | 4474 | 0.41 | 0.41–0.82 | 10 | 16 | 29 | 153 (103–202) | 16.27 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 |
| 3′ | 10 | 4595 | 4628 | 0.57 | 0.24–0.97 | 10 | 24 | 41 | 217 (175–258) | 23.98 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 | |
| 3′ lng1 | 7 | 4595 | 4616 | 0.41 | 0.24–0.55 | 7 | 18 | 25 | 160 (131, 189) | 17.62 | 0.8246 | Yes | No | Selection | Lng 1 | |
| 270015 | 5′ | 7 | 4449 | 4449 | 0.10 | 0–0.27 | 7 | 4.6 | 12 | 43 (6, 81) | 4.571 | 1.80 × 10−5 | No | Yes | ≥2 | |
| 3′ | 6 | 4622 | 4623 | 0.16 | 0–0.46 | 6 | 8 | 21 | 73 (22, 167) | 8 | 2.00 × 10−16 | No | Yes | ≥2 | ||
| 3′ lng1 | 5 | 4622 | 4623 | 0.03 | 0–0.07 | 5 | 2 | 3 | 18 (8, 29) | 2 | 0.6198 | Yes | Yes | Lng 1 | ||
| 192023 | 5′ | 6 | 4448 | 4448 | 0.48 | 0.16–0.99 | 6 | 19 | 34 | 184 (80, 288) | 19.47 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 |
| 3′ | 6 | 4604 | 4607 | 0.54 | 0.17–0.72 | 6 | 23 | 32 | 206 (154, 258) | 22.6 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 | |
| 193004 | 5′ | 8 | 4460 | 4494 | 0.87 | 0.36–1.41 | 8 | 36 | 60 | 340 (242, 439) | 36.46 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 |
| 3′ | 8 | 4636 | 4673 | 1.67 | 0.51–3.39 | 8 | Too high | No | No | ≥2 | ||||||
| 193005 | 5′ | 6 | 4482 | 4485 | 1.22 | 0.31–2.30 | 6 | 49 | 87 | 456 (187, 724) | 48.67 | 2.00 × 10−16 | No | No | Multiple strains | ≥2 |
| 3′ | 6 | 4469 | 4678 | 0.20–11.01 | 6 | Too high | No | No | Multiple strains | ≥2 | ||||||
| 191696 | 5′ | 3 | 4450 | 4450 | 0.64 | 0.34–0.89 | 3 | N/A | No | No | Multiple strains | ≥2 | ||||
| 3′ | 3 | 4575 | 4602 | 0.54 | 0.13–0.77 | 3 | N/A | No | No | Multiple strains | ≥2 | |||||
| 191997 | 5′ | 9 | 4466 | 4468 | 0.20 | 0.02–0.33 | 9 | 5.8 | 13 | 62 (42, 83) | 5.778 | 0.00941 | No | No (f) | Selection | ≥2 |
| 3′ | 10 | 4651 | 4664 | 0.25 | 0–0.41 | 10 | 11 | 18 | 97 (75, 119) | 10.78 | 2.00 × 10−16 | No | No | Selection | ≥2 | |
| 194037 | 5′ | 12 | 4456 | 4456 | 0.04 | 0–0.11 | 12 | 1.7 | 5 | 16 (9, 23) | 1.667 | 0.9321 | Yes | Yes | 1 | |
| 3′ | 12 | 4596 | 4599 | 0.06 | 0–0.15 | 12 | 2.6 | 7 | 24 (14, 34) | 2.636 | 0.9332 | Yes | Yes | 1 | ||
| 194584 | 5′ | 14 | 4455 | 4456 | 0.08 | 0–0.20 | 14 | 1.6 | 4 | 17 (11, 23) | 1.571 | 0.7648 | Yes | Yes (f) | 1 | |
| 3′ | 7 | 4580 | 4580 | 0.07 | 0.02–0.15 | 7 | 3.3 | 7 | 31 (15, 46) | 3.333 | 0.9464 | Yes | Yes | 1 | ||
| 270535 | 5′ | 8 | 4459 | 4460 | 0.11 | 0.04–0.18 | 8 | 4.9 | 8 | 47 (34, 59) | 4.929 | 0.6833 | Yes | Yes | 1 | |
| 3′ | 5 | 4576 | 4601 | 0.11 | 0.09–0.13 | 5 | 5.2 | 6 | 48 (40,56) | 5.2 | 0.3723 | Yes | Yes | 1 | ||
| 191639 | 5′ | 6 | 4458 | 4459 | 0.04 | 0–0.09 | 6 | 2 | 4 | 19 (7, 31) | 2 | 0.7987 | Yes | Yes | 1 | |
| 3′ | 8 | 4618 | 4619 | 0.19 | 0.07–0.43 | 8 | 3.6 | 8 | 38 (20, 56) | 3.607 | 0.8119 | Yes | yes (f) | 1 | ||
| 194603 | 5′ | 6 | 4461 | 4461 | 0.10 | 0–0.22 | 6 | 3 | 7 | 33 (5, 60) | 3 | 0.8227 | Yes | Yes (f) | 1 | |
| 3′ | 6 | 4623 | 4626 | 0.19 | 0.11–0.22 | 6 | 5.3 | 7 | 55 (40, 70) | 5.333 | 0.8391 | Yes | yes (f) | 1 | ||
| 275031 | 5′ | 4 | 4547 | 4465 | 0.04 | 0.02–0.07 | 4 | 2 | 3 | 19 (4, 33) | 2 | 0.6822 | Yes | Yes | 1 | |
| 3′ | 6 | 4605 | 4606 | 0.07 | 0.02–0.13 | 6 | 2.3 | 4 | 24 (13, 35) | 2.333 | 0.5462 | Yes | yes (f) | 1 | ||
| 194604 | 5′ | 10 | 4459 | 4459 | 0.03 | 0–0.09 | 10 | 1.2 | 4 | 11 (4, 19) | 1.2 | 0.5375 | Yes | Yes | 1 | |
| 3′ | 10 | 4575 | 4575 | 0.07 | 0.02–0.18 | 10 | 3.3 | 8 | 30 (17, 40) | 3.289 | 0.7812 | Yes | No | Selection | 1 | |
| 192002 | 5′ | 4 | 4191 | 4477 | 0.16 | 0.07–0.26 | 4 | 7 | 11 | 66 (22, 110) | 7 | 0.9283 | Yes | Yes | 1 | |
| 3′ | 8 | 4549 | 4595 | 0.17 | 0.02–0.33 | 8 | 8.2 | 15 | 75 (48, 102) | 8.214 | 0.898 | Yes | No | Selection | 1 | |
| 194535 | 5′ | 7 | 4459 | 4459 | 0.08 | 0.02–0.11 | 7 | 3.4 | 5 | 32 (21, 44) | 3.429 | 0.6776 | Yes | Yes | 1 | |
| 3′ | 7 | 4624 | 4818 | 0.17 | 0.11–0.30 | 6 | 5.8 | 8 | 51 (43, 58) | 5.8 | 0.6561 | Yes | No (f) | Selection | 1 | |
| 194319 | 5′ | 7 | 4461 | 4461 | 0.05 | 0–0.09 | 7 | 2.3 | 4 | 22 (17, 26) | 2.286 | 0.7703 | Yes | No | Early selection | 1 |
| 3′ | 7 | 4575 | 4593 | 0.10 | 0.07–0.13 | 7 | 4.4 | 6 | 40 (32, 48) | 4.381 | 0.4586 | Yes | No | Early selection | 1 | |
| 193006 | 5′ | 6 | 4452 | 4452 | 0.07 | 0.02–0.11 | 6 | 3 | 5 | 28 (15, 42) | 3.0 | 0.8046 | Yes | Yes | 1 | |
| 3′ | 6 | 4435 | 4489 | 0.11 | 0.04–0.18 | 6 | 5.2 | 8 | 49 (29, 68) | 5.2 | 0.4767 | Yes | No | Early stochastic | 1 | |
| 194020 | 5′ | 5 | 4463 | 4463 | 0.06 | 0–0.13 | 5 | 2.8 | 6 | 27 (-3, 56) | 2.8 | 0.6693 | Yes | Yes | 1 | |
| 3′ | 5 | 4623 | 4623 | 0.19 | 0.14–0.22 | 5 | 8.5 | 10 | 77 (68, 87) | 8.5 | 0.4971 | Yes | No | Selection | 1 | |
| 194346 | 5′ | 5 | 4460 | 4460 | 0.06 | 0–0.11 | 5 | 2.8 | 5 | 27 (9, 44) | 2.8 | 0.7886 | Yes | Yes | 1 | |
| 3′ | 5 | 4599 | 4602 | 0.14 | 0.11–0.20 | 5 | 6.8 | 10 | 62 (44, 80) | 6.8 | 0.8729 | Yes | No | Selection | 1 | |
| 194374 | 5′ | 7 | 4455 | 4458 | 0.09 | 0–0.16 | 7 | 3.8 | 7 | 36 (24, 49) | 3.81 | 0.7509 | Yes | No | Early selection | 1 |
| 3′ | 6 | 4638 | 4638 | 0.07 | 0–0.17 | 6 | 3.3 | 8 | 30 (10, 50) | 3.267 | 0.657 | Yes | No | Early selection | 1 | |
| 191955 | 5′ | 6 | 4473 | 4475 | 0.10 | 0.07–0.13 | 6 | 4.7 | 6 | 44 (33, 55) | 4.667 | 0.4987 | Yes | Yes | 1 | |
| 3′ | 6 | 4630 | 4642 | 0.11 | 0–0.26 | 6 | 5 | 12 | 45 (6, 85) | 5 | 0.00015 | No | Yes | 1 | ||
| 191923 | 5′ | 11 | 4461 | 4461 | 0.11 | 0–0.36 | 11 | 1.8 | 6 | 20 (9, 31) | 1.818 | 0.3695 | Yes | Yes | 1 | |
| 3′ | 11 | 4490 | 4613 | 0.12 | 0–0.24 | 11 | 5.2 | 11 | 47 (30, 64) | 5.164 | 2.08 × 10−13 | No | No | Selection | 1 | |
| 194065 | 5′ | 11 | 4459 | 4459 | 0.06 | 0–0.16 | 11 | 2.9 | 7 | 28 (14, 41) | 2.909 | 0.4872 | Yes | Yes | 1 | |
| 3′ | 11 | 4591 | 4594 | 0.22 | 0–0.33 | 11 | 9.2 | 15 | 84 (68, 101) | 9.2 | 0.00063 | No | No | Selection | 1 | |
| 194289 | 5′ | 11 | 4425 | 4446 | 0.05 | 0–0.20 | 11 | 2.2 | 9 | 21 (6, 35) | 2.182 | 1.23 × 10−8 | No | Yes | yes | 1 |
| 3′ | 9 | 4599 | 4614 | 0.18 | 0–0.26 | 9 | 7.6 | 11 | 70 (57, 82) | 7.639 | 0.00011 | No | No | Selection | 1 | |
| 193008 | 5′ | 5 | 4491 | 4492 | 0.09 | 0.04–0.16 | 5 | 4.2 | 7 | 39 (21, 58) | 4.2 | 0.9419 | Yes | No | Early stochastic | 1 |
| 3′ | 6 | 4667 | 4670 | 0.18 | 0.02–0.29 | 6 | 8.7 | 13 | 78 (53, 103) | 8.667 | 0.00063 | No | No | Selection | 1 | |
| 275027 | 5′ | 9 | 4455 | 4455 | 0.15 | 0–0.29 | 9 | 6.4 | 12 | 60 (42, 79) | 6.389 | 2.00 × 10−16 | No | No | Selection | 1 |
| 3′ | 2 | 4302 | 4641 | 0.26 | N/A | 2 | N/A | ? | ||||||||
| 191996 | 5′ | 4 | 4459 | 4461 | 0.51 | 0.04–0.75 * | 4 | N/A. Two out of four sequences were hypermutated by APOBEC3G (A3G) | N/A | A3G Hypermutation | ? | |||||
| 3′ | 3 | 4633 | 4634 | 0.13 | 0.11–0.15 | 3 | N/A | Yes | ||||||||
| 194140 | 5′ | 2 | 4457 | 4457 | 0 | N/A | 2 | N/A | ? | |||||||
| 3′ | 2 | 4677 | 4677 | 0.02 | 2 | |||||||||||
| Unique Identifier | No. of Break Points | Breakpoint Interval | Length, No. Nucleotides | Directionality of Recombination | Envelope Region of Recombination Events | Recombination Hotspot # | vpu | Subtype Composition of Overlapping vpu, tat and Rev Genes/Exons | |
|---|---|---|---|---|---|---|---|---|---|
| Start–End, HXB2 Numbering | tat-1, rev-1 | tat-2, rev-2 | |||||||
| 194319-env | 1 | 8214–8252 | 38 | A1/D | TM domain, gp41 | #1 | A1/D | D | D |
| 194140-env | 1 | 8251–8313 | 62 | A1/D | TM domain, gp41 | #1 | A1 | D | D |
| 194065-env | 1 | 8258–8358 | 100 | A1/D | TM domain, gp41 | #1 | A1 | D | D |
| 191696-env | 1 | 8251–8303 | 52 | A1/D | TM domain, gp41 | #1 | A1/D | D | D |
| 194584-env | 2 | 6990–7202 | 212 | D/A1 | V3, CD4/co-receptor binding | D | D | D | |
| 8252–8330 | 78 | A1/D | TM domain, gp41 | #1 | |||||
| 194020-env | 2 | 6975–7057 | 82 | D/A1 | CD4 contact, near V3 | D | D | D | |
| 8252–8301 | 49 | A1/D | TM domain, gp41 | #1 | |||||
| 191997-env | 2 | 7116–7135 | 19 | D/A1 | V3 loop | D | D | D | |
| 8213–8305 | 92 | A1/D | TM domain, gp41 | #1 | |||||
| 191996-env | 2 | 6336–6364 | 28 | D/A1 | Signal peptide/C1 | #2 | D | D | D |
| 8272–8299 | 27 | A1/D | TM domain, gp41 | #1 | |||||
| 275026-env | 3 | 6326–6360 | 34 | D/A1 | Signal peptide/C1 | #2 | D | D | D |
| 8270–8333 | 63 | A1/D | TM domain, gp41 | #1 | |||||
| 8722–8740 | 18 | D/A1 | LLP-1 | ||||||
| 194603-env | 3 | 7329–? | ? | A1/D | CD4 binding | A1 | D | D | |
| 7473–? | ? | D/A1 | Coreceptor binding | ||||||
| 8386–8399 | 13 | A1/D | TM domain, gp41 | #1 | |||||
| 270015-env | 4 | 6309–6351 | 42 | D/A1 | Signal peptide/C1 | #2 | D | D | D |
| 6622–6674 | 52 | A1/D | V1 loop | ||||||
| 7470–7506 | 36 | D/A1 | CD4/coreceptor binding | ||||||
| 8251–8297 | 46 | A1/D | TM domain, gp41 | #1 | |||||
| 194346-env | 4 | 6339–6365 | 26 | D/A1 | Signal peptide/C1 | #2 | D | D | D |
| 6427–6468 | 41 | A1/D | Signal peptide/C1 | #2 | |||||
| 6745–6779 | 34 | D/A1 | V2 | ||||||
| 8246–8296 | 50 | A1/D | TM domain, gp41 | #1 | |||||
| 193006-env | 1 | 8031–8074 | 43 | A1/D | gp41 fusion domain | A1/D | D | D | |
| 193005-env | 2 | 7313–7327 | 14 | D/A1 | CD4 binding loop | D | D | D | |
| 7519–7540 | 21 | A1/ D | Coreceptor binding | ||||||
| 193004-env | 2 | 6426–6475 | 49 | D/A1 | Signal peptide/C1 | #2 | D | D | D |
| 6795–6889 | 94 | A1/ D | V2 loop | ||||||
| 191923-env | 2 | 7018–7055 | 37 | D/A1 | CD4 contact, near V3 | D | D | D | |
| Env Subtype | Amino Acid (Single Nucleotide Code) | Sample ID |
|---|---|---|
| D | H | 194604 |
| D | H | 194535 |
| D | H | 192023 |
| D | H | 191923 |
| D | H | 194020 |
| D | H | 270015 |
| D | H | 191997 |
| D | H | 275031 |
| D | H | 191996 |
| D | H | 194307 |
| D | H | 194374 |
| D | N/A | 193004 |
| D | N/A | 193005 |
| D | Q | 192002 |
| D | Q | 275027 |
| D | G | 194289 |
| D | I | 194584 |
| D | L | 270535 |
| D | T/P | 275026 |
| D | Del | 191639 |
| D | D | 191955 |
| A1 | N | 193006 |
| A1 | N | 194603 |
| A1 | N | 191696 |
| A1 | N | 194319 |
| A1 | N | 194140 |
| A1 | D | 194346 |
| A1 | D | 194065 |
| A1 | Y | 193008 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balinda, S.N.; Kapaata, A.; Xu, R.; Salazar, M.G.; Mezzell, A.T.; Qin, Q.; Herard, K.; Dilernia, D.; Kamali, A.; Ruzagira, E.; et al. Characterization of Near Full-Length Transmitted/Founder HIV-1 Subtype D and A/D Recombinant Genomes in a Heterosexual Ugandan Population (2006–2011). Viruses 2022, 14, 334. https://doi.org/10.3390/v14020334
Balinda SN, Kapaata A, Xu R, Salazar MG, Mezzell AT, Qin Q, Herard K, Dilernia D, Kamali A, Ruzagira E, et al. Characterization of Near Full-Length Transmitted/Founder HIV-1 Subtype D and A/D Recombinant Genomes in a Heterosexual Ugandan Population (2006–2011). Viruses. 2022; 14(2):334. https://doi.org/10.3390/v14020334
Chicago/Turabian StyleBalinda, Sheila N., Anne Kapaata, Rui Xu, Maria G. Salazar, Allison T. Mezzell, Qianhong Qin, Kimberly Herard, Dario Dilernia, Anatoli Kamali, Eugene Ruzagira, and et al. 2022. "Characterization of Near Full-Length Transmitted/Founder HIV-1 Subtype D and A/D Recombinant Genomes in a Heterosexual Ugandan Population (2006–2011)" Viruses 14, no. 2: 334. https://doi.org/10.3390/v14020334