
Genome-Wide Analysis of Differentially Expressed mRNAs and lncRNAs in Koi Carp Infected with Koi Herpesvirus
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. RNA Extraction, Library Construction, and Sequencing
2.3. Identification, Annotation, Prediction, and Structural Analysis of lncRNAs
2.4. Differential Expression and Functional Enrichment Analyses
2.5. LncRNA–mRNA Network Analysis
2.6. Validation of the RNA-Sequencing Results via Quantitative PCR
3. Results
3.1. Quality and Validation of Transcriptomic Data
3.2. Identification of lncRNAs in Koi Carp Tissues
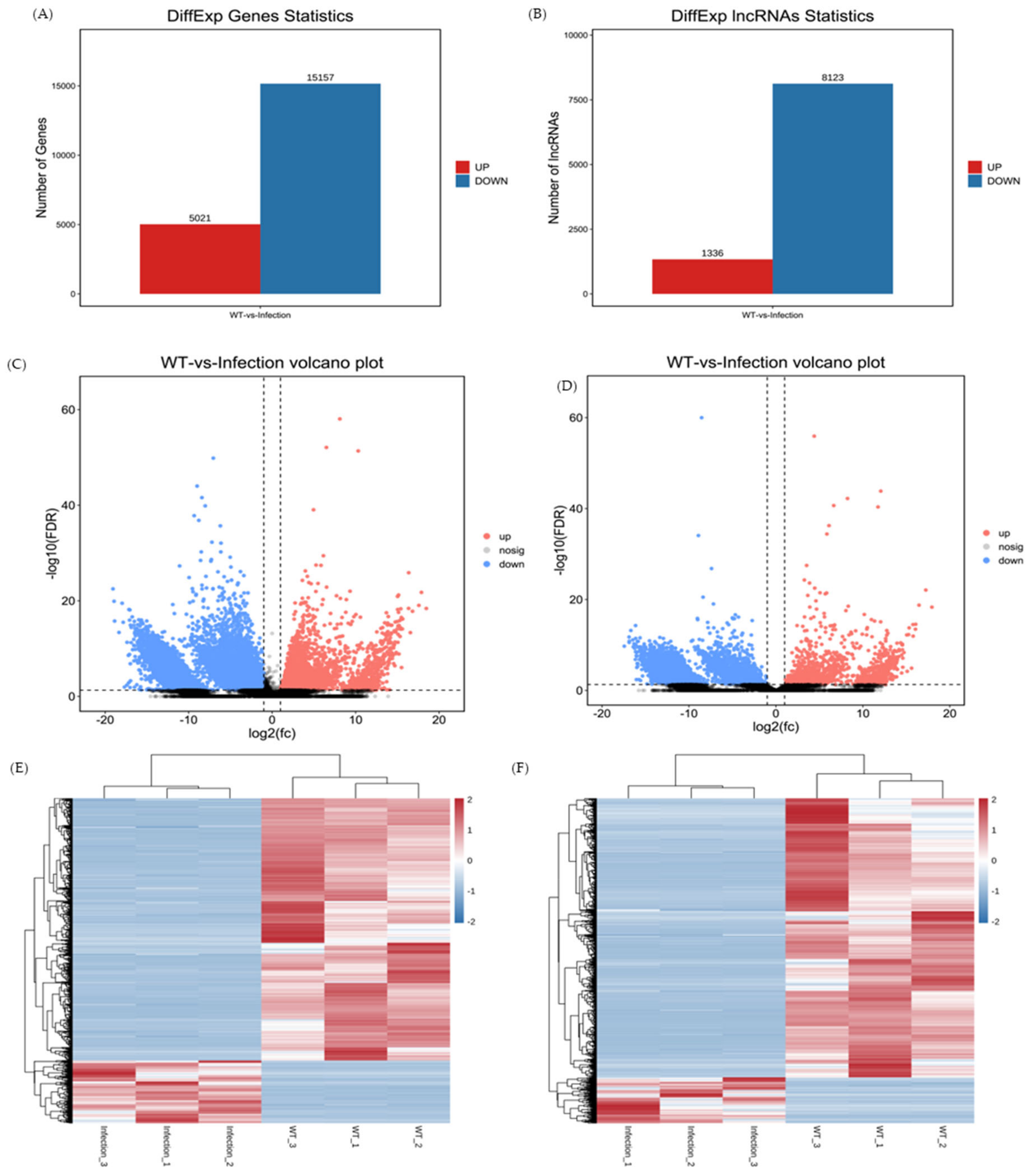
3.3. Profiles of DEmRNAs and DElncRNAs
3.4. GO Analyses of the DEGs
3.5. KEGG Analyses of the DEGs
3.6. Prediction and Functional Analysis of mRNA Targets of DElncRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Ethics Statement
References
- Rinn, J.L.; Chang, H.Y. Genome Regulation by Long Noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [Green Version]
- Batista, P.J.; Chang, H.Y. Long Noncoding RNAs: Cellular Address Codes in Development and Disease. Cell 2013, 152, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cech, T.R.; Steitz, J.A. The Noncoding RNA Revolution-Trashing Old Rules to Forge New Ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and Functions of Long Noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Hu, J.Y.; Chen, J.L. lncRNAs regulate the innate immune response to viral infection. Wiley Interdiscip. Rev.-RNA 2016, 7, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Gralinski, L.; Armour, C.D.; Ferris, M.T.; Thomas, M.J.; Proll, S.; Bradel-Tretheway, B.G.; Korth, M.J.; Castle, J.C.; Biery, M.C.; et al. Unique Signatures of Long Noncoding RNA Expression in Response to Virus Infection and Altered Innate Immune Signaling. Mbio 2010, 1, e00206-10. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jiang, S.; Wu, W.; Yu, F.; Chang, W.G.; Li, P.F.; Wang, K. Non-coding RNAs Function as Immune Regulators in Teleost Fish. Front. Immunol. 2018, 9, 2801. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hu, Z.; Zhang, Y.; Wang, C. Long non-coding RNAs in Epstein-Barr virus-related cancer. Cancer Cell Int. 2021, 21, 278. [Google Scholar] [CrossRef]
- Robinson, E.K.; Covarrubias, S.; Carpenter, S. The how and why of lncRNA function: An innate immune perspective. Biochim. Et Biophys. Acta-Gene Regul. Mech. 2020, 1863, 194419. [Google Scholar] [CrossRef]
- Serpeloni, J.M.; Neto, Q.A.; Lucio, L.C.; Ramao, A.; de Oliveira, J.C.; Gradia, D.F.; Malheiros, D.; Ferrasa, A.; Marchi, R.; Figueiredo, D.L.; et al. Genome interaction of the virus and the host genes and non-coding RNAs in SARS-CoV-2 infection. Immunobiology 2021, 226, 152130. [Google Scholar] [CrossRef]
- Wang, J.; Cen, S. Roles of lncRNAs in influenza virus infection. Emerg. Microbes Infect. 2020, 9, 1407–1414. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhang, N.; Chen, Y.; Yin, J.; Xu, M.; Cheng, X.; Ma, R.; Meng, J.; Du, Y. Role of Non-Coding RNA in Neurological Complications Associated with Enterovirus 71. Front. Cell. Infect. Microbiol. 2022, 12, 873304. [Google Scholar] [CrossRef] [PubMed]
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adamek, M.; Steinhagen, D.; Irnazarow, I.; Hikima, J.; Jung, T.S.; Aoki, T. Biology and host response to Cyprinid herpesvirus 3 infection in common carp. Dev. Comp. Immunol. 2014, 43, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Gotesman, M.; Kattlun, J.; Bergmann, S.M.; El-Matbouli, M. CyHV-3: The third cyprinid herpesvirus. Dis. Aquat. Org. 2013, 105, 163–174. [Google Scholar] [CrossRef] [Green Version]
- Jia, Z.; Wu, N.; Jiang, X.; Li, H.; Sun, J.; Shi, M.; Li, C.; Ge, Y.; Hu, X.; Ye, W.; et al. Integrative Transcriptomic Analysis Reveals the Immune Mechanism for a CyHV-3-Resistant Common Carp Strain. Front. Immunol. 2021, 12, 687151. [Google Scholar] [CrossRef]
- Song, F.; Wang, L.; Zhu, W.; Dong, Z. Long noncoding RNA and mRNA expression profiles following igf3 knockdown in common carp, Cyprinus carpio. Sci. Data 2019, 6, 190024. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- Kang, Y.J.; Yang, D.C.; Kong, L.; Hou, M.; Meng, Y.Q.; Wei, L.P.; Gao, G. CPC2: A fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017, 45, W12–W16. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Araki, M.; Goto, S.; Hattori, M.; Hirakawa, M.; Itoh, M.; Katayama, T.; Kawashima, S.; Okuda, S.; Tokimatsu, T.; et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M. Activation and Evasion of Antiviral Innate Immunity by Hepatitis C Virus. J. Mol. Biol. 2014, 426, 1198–1209. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.Z.; Ma, J.Y.; Yoo, D. Inhibition of NF-kappa B activity by the porcine epidemic diarrhea virus nonstructural protein 1 for innate immune evasion. Virology 2017, 510, 111–126. [Google Scholar] [CrossRef]
- Zhang, Q.Z.; Yoo, D.W. Immune evasion of porcine enteric coronaviruses and viral modulation of antiviral innate signaling. Virus Res. 2016, 226, 128–141. [Google Scholar] [CrossRef]
- West, J.A.; Gregory, S.M.; Sivaraman, V.; Su, L.S.; Damania, B. Activation of Plasmacytoid Dendritic Cells by Kaposi's Sarcoma-Associated Herpesvirus. J. Virol. 2011, 85, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Gregory, S.M.; West, J.A.; Dillon, P.J.; Hilscher, C.; Dittmer, D.P.; Damania, B. Toll-like receptor signaling controls reactivation of KSHV from latency. Proc. Natl. Acad. Sci. USA 2009, 106, 11725–11730. [Google Scholar] [CrossRef] [Green Version]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.I.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a Viral NLR Homolog that Inhibits the Inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef]
- West, J.A.; Wicks, M.; Gregory, S.M.; Chugh, P.; Jacobs, S.R.; Zhang, Z.; Host, K.M.; Dittmer, D.P.; Damania, B. An important role for mitochondrial antiviral signaling protein in the Kaposi's sarcoma-associated herpesvirus life cycle. J. Virol. 2014, 88, 5778–5787. [Google Scholar] [CrossRef]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Ulitsky, I.; Bartel, D.P. lincRNAs: Genomics, Evolution, and Mechanisms. Cell 2013, 154, 26–46. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular Mechanisms of Long Noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Kino, T.; Hurt, D.E.; Ichijo, T.; Nader, N.; Chrousos, G.P. Noncoding RNA Gas5 Is a Growth Arrest- and Starvation-Associated Repressor of the Glucocorticoid Receptor. Sci. Signal. 2010, 3, ra8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long Noncoding RNA as Modular Scaffold of Histone Modification Complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brockdorff, N. Noncoding RNA and Polycomb recruitment. RNA 2013, 19, 429–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Chu, Q.; Xu, T.J.; Zheng, W.W.; Chang, R.J.; Zhang, L. Long noncoding RNA MARL regulates antiviral responses through suppression miR-122-dependent MAVS downregulation in lower vertebrates. PLoS Pathog. 2020, 16, e1008670. [Google Scholar] [CrossRef]
- Zheng, W.; Chu, Q.; Xu, T. Long noncoding RNA IRL regulates NF-κB-mediated immune responses through suppression of miR-27c-3p-dependent IRAK4 downregulation in teleost fish. J. Biol. Chem. 2021, 296, 100304. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.W.; Chu, Q.; Xu, T.J. The long noncoding RNA NARL regulates immune responses via microRNA-mediated NOD1 downregulation in teleost fish. J. Biol. Chem. 2021, 296, 100414. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′to 3′) |
|---|---|
| 31942.2-qpcr-F | TGACTGGGAGTGATGGTTTA |
| 31942.2-qpcr-R | AGAATGCCTTGAGGGTGAGT |
| 84010.1-qpcr-F | CCACCTAGTGGTCAGGAGAT |
| 84010.1-qpcr-R | GACCCACCGAATACAAAGAG |
| 837.2-qpcr-F | CCCGAGAATCGTTCTGTAGTGG |
| 837.2-qpcr-R | TCTGGGCCTAATGCTGTCAATC |
| 84012.1-qpcr-F | TGACCCACCGAATACAAAGA |
| 84012.1-qpcr-R | GAGGCTATAACTCCGCAACG |
| 9580.5-qpcr-F | GGGGATTCTCCTTGCTGTAT |
| 9580.5-qpcr-R | GGGCCATCTATCTTCTTTGC |
| 4779.1-qpcr-F | TTTATTTAAGGGTGGCAGTG |
| 4779.1-qpcr-R | TTCTTTGAGGGTTTGTTTCA |
| 5436.4-qpcr-F | AACAACTGCCGCATAATGAAAC |
| 5436.4-qpcr-R | AGCAACCATAGACCAGCAACAA |
| 15250.1-qpcr-F | AAGTGAACTGTATGGCAGAG |
| 15250.1-qpcr-R | ATGAAAGAATGAACGGATGA |
| 40348.1-qpcr-F | ATTTGTTGGTGCTGTTGTCC |
| 40348.1-qpcr-R | TGAGGCTTTACGCATAGGTT |
| 40352.1-qpcr-F | ATTTGTTGGTGCTGTTGTCC |
| 40352.1-qpcr-R | AGGCTTTACGCATAGGTTGT |
| 44075.1-qpcr-F | CGCATTTCATAATATCCACC |
| 44075.1-qpcr-R | ATAACACCAGAAGCAGACCA |
| 52488.1-qpcr-F | GAGTCGTTGCTATCGCTTGG |
| 52488.1-qpcr-R | ACCTCTGCCTCATCTTTCTC |
| 52508.1-qpcr-F | AGGAGAAAGATGAGGCAGAG |
| 52508.1-qpcr-R | TGAGGAGATTGAAGTGGAGG |
| 64495.2-qpcr-F | CCACCTTGCGACTAAATTCTAAATC |
| 64495.2-qpcr-R | TACAAAATTGAAAGCCAGTCACAAC |
| 65064.1-qpcr-F | GCGGTGTTCTTCTGTCTGCA |
| 65064.1-qpcr-R | GCGTCGGGTCTTGGGTTCTA |
| 73364.3-qpcr-F | TGCTGGGAGGCTGAGGCTGG |
| 73364.3-qpcr-R | CGCGGACACCCGATCGACAT |
| 76443.1-qpcr-F | CCTTCGCCCTTGTTTCGGTG |
| 76443.1-qpcr-R | AGACACGGGAGTCGGGAGGC |
| 80990.1-qpcr-F | CCTCTTAATTGAATGCCCCTCCTC |
| 80990.1-qpcr-R | TCTCCGTCATAGCGGTTTTACTTG |
| 87639.1-qpcr-F | TCAGTAAATTCGATTTTAATGTGGG |
| 87639.1-qpcr-R | ATTGAAGTATTTTCTATCCTGTGGC |
| 91318.1-qpcr-F | GGGGCAACAGCTTCTAATGT |
| 91318.1-qpcr-R | CCAGCCACCAAAGACAGATT |
| BAD-QPCR-F | ATGTGCGTGGAAAGCGTCAAC |
| BAD-QPCR-R | AAAGGCTCCGATGGTCACTCC |
| Bax-qpcr-F | GGCTATTTCAACCAGGGTTCC |
| Bax-qpcr-R | TGCGAATCACCAATGCTGT |
| MYD88-QPCR-F | AGCCTTTGCCCAGGAACTC |
| MYD88-QPCR-R | TGTGTGGAGGGTCTGGTGTA |
| TRAF6-QPCR-F | CCGGACCGAAACAGTATAATGGC |
| TRAF6-QPCR-R | CCACGTCATAGCCTTGCTGA |
| IRF-QPCR-3F | CAGGCATACGGAGGACATT |
| IRF-QPCR-3R | TGGCTTCAGGTCTGTTTTTG |
| ATG16L-qpcr-F | ATCAGGTCGGAGAGTATCGT |
| ATG16L-qpcr-R | GTTTTGTCTAGTTTCCCCGT |
| MAVS-QPCR-F | TCACACTCACTGATAGGGAAGAG |
| MAVS-QPCR-R | TAGCTCTCATCTCATTAGCCAGT |
| TBK1-QPCR-F | AGAGGAGTATCTGCATCCTGAC |
| TBK1-QPCR-R | CCTTCGTACGGTCTGAACG |
| RELA-QPCR-F | CACTGTAACTGTGTGTGTTTGTCT |
| RELA-QPCR-R | CCGCTGTAGTTGTGAACCTTGA |
| IFN-1-QPCR-F | ACCAGGTGAGGTTTCTTGTC |
| IFN-1-QPCR-R | CCACTGTCGTTAGGTTCCAT |
| Gene | Sequencing Result (Infection vs. NC) | Real-Time PCR Validation Result (Infection vs. NC) | Consistent or Not |
|---|---|---|---|
| 837.2 | - | - | Y |
| 4779.1 | - | - | Y |
| 5436.4 | - | - | Y |
| 9580.5 | - | - | Y |
| 15250.1 | - | - | Y |
| 31942.2 | - | - | Y |
| 40348.1 | - | - | Y |
| 40352.1 | - | - | Y |
| 44075.1 | - | - | Y |
| 52488.1 | - | - | Y |
| 52508.1 | - | - | Y |
| 64495.2 | - | - | Y |
| 65064.1 | - | - | Y |
| 73364.3 | + | + | Y |
| 76443.1 | - | - | Y |
| 80990.1 | - | - | Y |
| 84010.1 | - | - | Y |
| 84012.1 | - | - | Y |
| 87639.1 | - | - | Y |
| 91318.1 | - | - | Y |
| BAD | - | - | Y |
| Bax | - | - | Y |
| Myd88 | - | - | Y |
| TRAF6 | - | - | Y |
| IRF3 | - | - | Y |
| ATG16L | - | - | Y |
| MAVS | - | - | Y |
| TBK1 | - | - | Y |
| RELA | - | - | Y |
| IFN1 | - | - | Y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Z.; Luo, W.; Huang, Z.; Guo, M.; He, X.; Fan, Z.; Wang, Q.; Qin, Q.; Yang, M.; Lee, X. Genome-Wide Analysis of Differentially Expressed mRNAs and lncRNAs in Koi Carp Infected with Koi Herpesvirus. Viruses 2022, 14, 2555. https://doi.org/10.3390/v14112555
Yang Z, Luo W, Huang Z, Guo M, He X, Fan Z, Wang Q, Qin Q, Yang M, Lee X. Genome-Wide Analysis of Differentially Expressed mRNAs and lncRNAs in Koi Carp Infected with Koi Herpesvirus. Viruses. 2022; 14(11):2555. https://doi.org/10.3390/v14112555
Chicago/Turabian StyleYang, Zimin, Wei Luo, Zhihong Huang, Min Guo, Xiaochuan He, Zihan Fan, Qing Wang, Qiwei Qin, Min Yang, and Xuezhu Lee. 2022. "Genome-Wide Analysis of Differentially Expressed mRNAs and lncRNAs in Koi Carp Infected with Koi Herpesvirus" Viruses 14, no. 11: 2555. https://doi.org/10.3390/v14112555